
Enantioselective syntheses of (R)-pipecolic acid, (2R,3R)-3-hydroxypipecolic acid, β-(+)-conhydrine and (−)-swainsonine using an aziridine derived common chiral synthon†
Subhash P. Chavan*,
Lalit B. Khairnar,
Kailash P. Pawar,
Prakash N. Chavan and
Sanket A. Kawale
CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pune-411008, Maharashtra, India. E-mail: sp.chavan@ncl.res.in
First published on 26th May 2015
Abstract
Concise total syntheses of (R)-pipecolic acid, (R)-ethyl-6-oxopipecolate, (2R,3R)-3-hydroxypipecolic acid and formal syntheses of β-(+)-conhydrine, (−)-lentiginosine, (−)-swainsonine and 1,2-di-epi-swainsonine have been accomplished starting from a common chiral synthon. The present strategy employs regioselective aziridine ring opening, Wittig olefination and RCM as the key chemical transformations.
Introduction
(S)-Pipecolic acid 1, is a cyclic non-proteinogenic amino acid1 and its natural and non natural derivatives have found widespread applications in therapeutic chemistry (Fig. 1). Pipecolic acid acts as an integral framework of many pharmaceutically important molecules such as immunosuppressors rapamycin,2 FK506, immunomycin,3 the antitumor antibiotic sandramycin,4 local anaesthetics like bupivacaine and ropivacaine.5 Similarly, (R)-pipecolic acid ent-1 is a key constituent of the histone deacetylase (HDAC) inhibitors recognized as potential anticancer drugs.6 (R)-Piperidin-2-ylmethanol 2, a reduced analogue of ent-1 has been used as a precursor in the synthesis of (−)-lentiginosine 6, a naturally occurring alkaloid having amylo-glucosidase enzyme inhibition properties.7 3-Hydroxypipecolic acids are important scaffolds with piperidine skeleton and found in many biologically significant molecules.8 Trans-(−)-(2R,3R)-3-hydroxypipecolic acid 3 is a cyclic β-hydroxy-α-amino acid that has been used as a precursor in the synthesis of (−)-swainsonine 4,9 a potent anti-cancer drug and specific inhibitor of α-D-mannosidase.10 The stereochemistry of enantiomer of 3 is found in (+)-febrifugine 10, a potent antimalarial agent,11 and its reduced analogues were found as important structure motifs of (+)-prosopinine 8 and (+)-prosophylline 9 which exhibit analgesic, anaesthetic and antibiotic activities.12 cis Stereoisomer of 3-hydroxypipecolic acid is main constituent of (+)-isofebrifugine 11 which also shows antimalarial activity and of tetrazomine 12 which shows antitumor antibiotic activities.13 Oxopipecolate 7 has been utilized in medicinal chemistry and to prepare an important class of antitumor agents and is useful for the synthesis of pipecolic acid derivatives (Fig. 1).14 Pipecolic acid and its 3-hydroxy derivatives have also been incorporated in peptides which can induce a β-turn with resultant therapeutic significance15 and are also useful as organocatalysts.16 In general, pipecolic acid and its diverse functionalities are useful chiral building blocks for the synthesis of a variety of pharmaceutically important molecules.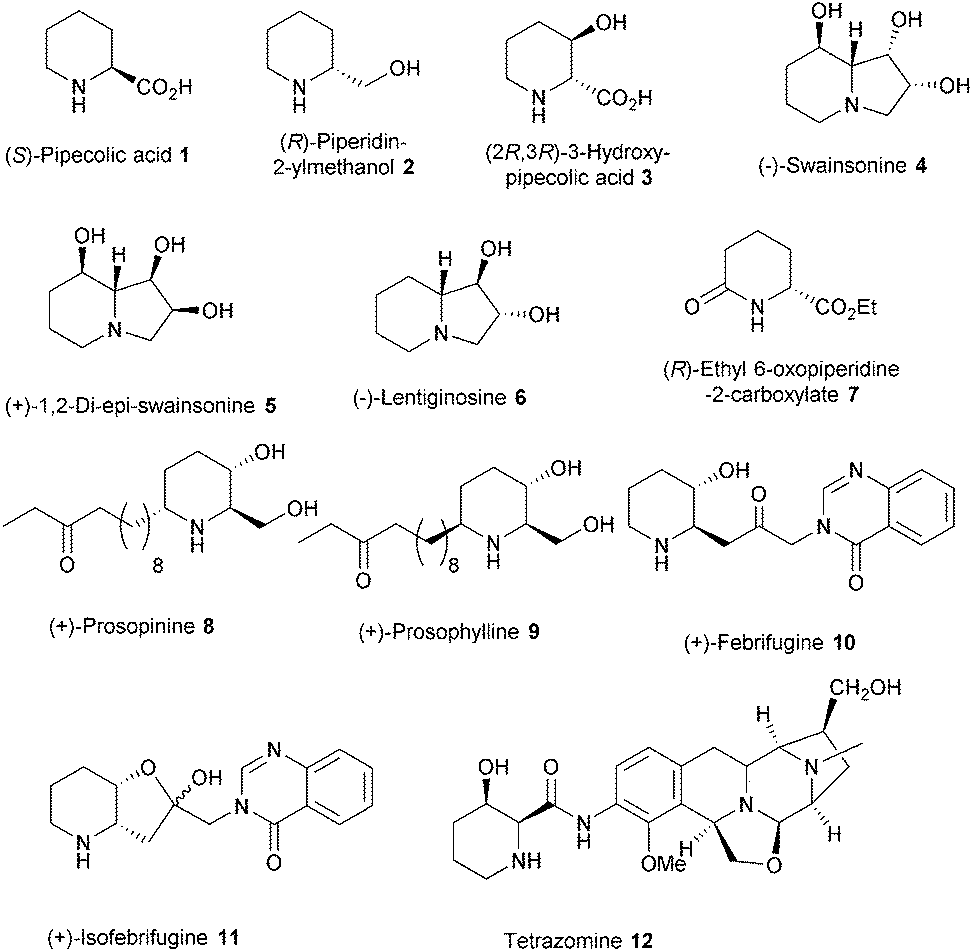 | ||
| Fig. 1 Pipecolic acid and its derivatives. | ||
Owing to their importance, enantiomerically pure syntheses of pipecolic acid and its derivatives has gained wide attention and have been extensively reviewed.17–19 As part of our continued interest toward development of efficient and practical synthetic routes to piperidine alkaloids,20 we report alternate practical and efficient syntheses of (R)-pipecolic acid ent-1, (R)-ethyl6-oxopipeclate 7, (2R,3R)-3-hydroxypipecolic acid 3 and formal synthesis of (−)-swainsonine 4, (+)-1,2-di-epi-swainsonine 5 and (−)-lentiginosine 6 from trans aziridine-2-carboxylate 13 as the common synthetic precursor derived from commercially available and cheap starting material viz. D-mannitol diacetonide. We have earlier described syntheses of (R) and (S) pipecolic acids and 3-hydroxypipecolic acid from cis aziridine.20
Results and discussion
Enantiomerically pure aziridines have been considered to be prominent precursors in the synthesis of natural and unnatural amino compounds due to their inherent ability to undergo regio and chemoselective nucleophilic ring opening reactions.21 Inspite of that, aziridines are relatively less explored compared to their three membered analog oxiranes due to less reactivity and selectivity towards ring opening reactions. However these drawbacks can be overcome by proper manipulations of functional groups attached to aziridine ring.Thus, as shown in the retrosynthetic plan (Scheme 1), it was envisioned that all of the six piperidine alkaloids could be derived from aziridine carboxylic acid 13 as the common precursor. For the synthesis of (R)-pipecolic acid (ent-1) as well as 3-hydroxypipecolic acid 3, it was surmised that vinyl aziridine ester 14 would serve as ideal substrate. The vinyl aziridine ester 14 could be readily derived from aziridine ester 13 by chain propagation from the acetonide side. Ester 14 by proper choice of reaction conditions could be converted to (R)-ethyl6-oxo-pipecolate 7, which serves as an important intermediate for pipecolic acid derivatives.14 Compound 7 in turn can be further converted to the intermediate 15, a precursor for the synthesis of (−)-lentiginosine22 and (R)-pipecolic acid ent-1. Aziridine ester 14 on regio and stereoselective ring opening under acidic condition by hydroxyl group could be converted to amino-alcohol 16 which could be further transformed to trans 3-hydroxypipecolic acid 3. Similarly, aziridine ester 13 on propagation from ester side would furnish α,β-unsaturated ester aziridine 17 which on regio and stereoselective ring opening by hydroxy group followed by cyclization, N-allylation and RCM would give intermediate 18, enantiomer of which is well explored towards the synthesis of (+)-swainsonine ent-4 and (−)-8,8a-di-epi-swainsonine ent-5.23
 | ||
| Scheme 1 Retrosynthetic analysis for target compounds. | ||
The synthetic endeavor for (R)-pipecolic acid ent-1 and derivatives started with readily available and cheap starting material D-mannitol diacetonide 19 (Scheme 2). Accordingly, D-mannitol diacetonide 19 on diol cleavage using NaIO4 yielded acetonide protected (R)-glyceraldehyde 20 which on Wittig olefination using bromophosphorane24 in dichloromethane afforded bromoester 21. The Gabriel–Cromwell reaction of 2-acrylic carboxylate derivative 21 with benzyl amine in toluene as solvent furnished trans aziridine-2-carboxylate 13 as the major isomer via conjugate addition, proton transfer and SN2 ring closure in 68% yield.25 The trans aziridine 13 was readily obtained by column chromatography. Acetonide trans aziridine 13 was deprotected using TMSOTf/CH2Cl2 at 0 °C,26 to furnish diol 22 in excellent yield (90%). Diol 22 was then subjected to sodium metaperiodate mediated oxidative cleavage to yield crude aldehyde which was subjected to Horner–Wadsworth–Emmons olefination to afford α,β-unsaturated aziridine-ester 14 in 70% yield over two steps. This α,β-unsaturated aziridine carboxylate 14 (Scheme 2) when subjected to palladium mediated transfer hydrogenation conditions,25b,27 underwent one- pot efficient regioselective aziridine ring cleavage with concomitant olefin reduction, N-debenzylation and cyclisation of resultant amine as the key step to give access to (R)-ethyl6-oxopipecolate 7 in 85% yield. Pipecolate 7 was further converted to (R)-N-Boc-2-piperidinemethanol 15 over two steps using LAH induced lactam/ester reduction followed by protection of resulting crude amino-alcohol as N-Boc derivative in 97% ee by HPLC analysis. (R)-N-Boc-2-piperidinemethanol 15 can be utilized as a precursor towards synthesis of (−)-lentiginosine 6.22 Finally, compound 15 was converted to (R)-pipecolic acid ent-1 by Boc deprotection using TFA followed by oxidation using KMnO4 in aqueous 3 N H2SO4. The synthesis of (R)-pipecolic acid has been achieved in 27% overall yield while (R)-ethyl6-oxo-pipecolate 7 was achieved in 54% overall yield from trans aziridine-2-carboxylate 13 respectively.
 | ||
| Scheme 2 Synthesis of (R)-pipecolic acid ent-1. | ||
After achieving the site selective functional group transformation at the acetonide group of aziridine-2-carboxylate 13, attention was focussed on regioselective ring opening of aziridine ring in compound 14 by water as the nucleophile. Accordingly, when compound 14 was treated with TFA (2 equiv.) in CH3CN–H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), it underwent regio and stereoselective nucleophilic ring opening reaction using water as nucleophile to afford γ-hydroxy-δ-amino-α,β-conjugated ester 16 as the only isomer in 76% yield (Scheme 3). Selective protection of hydroxyl group of amino-alcohol 16 was achieved using TBSCl, imidazole and cat. DMAP in refluxing dichloromethane to furnish TBS ether 23 in 85% yield. The crucial step viz. reductive cyclization of 23 was carried out under hydrogenation conditions using hydrogen gas and palladium hydroxide over carbon in ethanol to provide amide 24 in 85% yield (Scheme 3). In next step, amido ester 24 was subjected to selective reduction of amide functionality using borane dimethyl sulfide complex in anhydrous THF to furnish the amino ester 25 in 78% yield. Finally, the global deprotection involving ester hydrolysis as well as TBS group deprotection of 25 was carried out in a single step using 6 N HCl to provide (2R,3R)-3-hydroxypipecolic acid 3 in 91% yield. The spectral data and optical rotation values of 3 thus obtained were in good agreement with the reported one.28
:1), it underwent regio and stereoselective nucleophilic ring opening reaction using water as nucleophile to afford γ-hydroxy-δ-amino-α,β-conjugated ester 16 as the only isomer in 76% yield (Scheme 3). Selective protection of hydroxyl group of amino-alcohol 16 was achieved using TBSCl, imidazole and cat. DMAP in refluxing dichloromethane to furnish TBS ether 23 in 85% yield. The crucial step viz. reductive cyclization of 23 was carried out under hydrogenation conditions using hydrogen gas and palladium hydroxide over carbon in ethanol to provide amide 24 in 85% yield (Scheme 3). In next step, amido ester 24 was subjected to selective reduction of amide functionality using borane dimethyl sulfide complex in anhydrous THF to furnish the amino ester 25 in 78% yield. Finally, the global deprotection involving ester hydrolysis as well as TBS group deprotection of 25 was carried out in a single step using 6 N HCl to provide (2R,3R)-3-hydroxypipecolic acid 3 in 91% yield. The spectral data and optical rotation values of 3 thus obtained were in good agreement with the reported one.28
 | ||
| Scheme 3 Synthesis of (2R,3R)-3-hydroxy pipecolic acid 3. | ||
In order to ascertain the chiral purity, the lactam 24 was subjected to reduction using lithium aluminum hydride in anhydrous THF followed by N-Boc protection to provide N-Boc derivative 26. The chiral HPLC analysis of the 26 revealed that the chiral purity was ∼97% ee (Scheme 4). Thus, the total synthesis of trans (2R,3R)-3-hydroxypipecolic acid 3 was accomplished in 24.6% overall yield in 8 steps from trans aziridine 13.
 | ||
| Scheme 4 Synthesis of diol 26. | ||
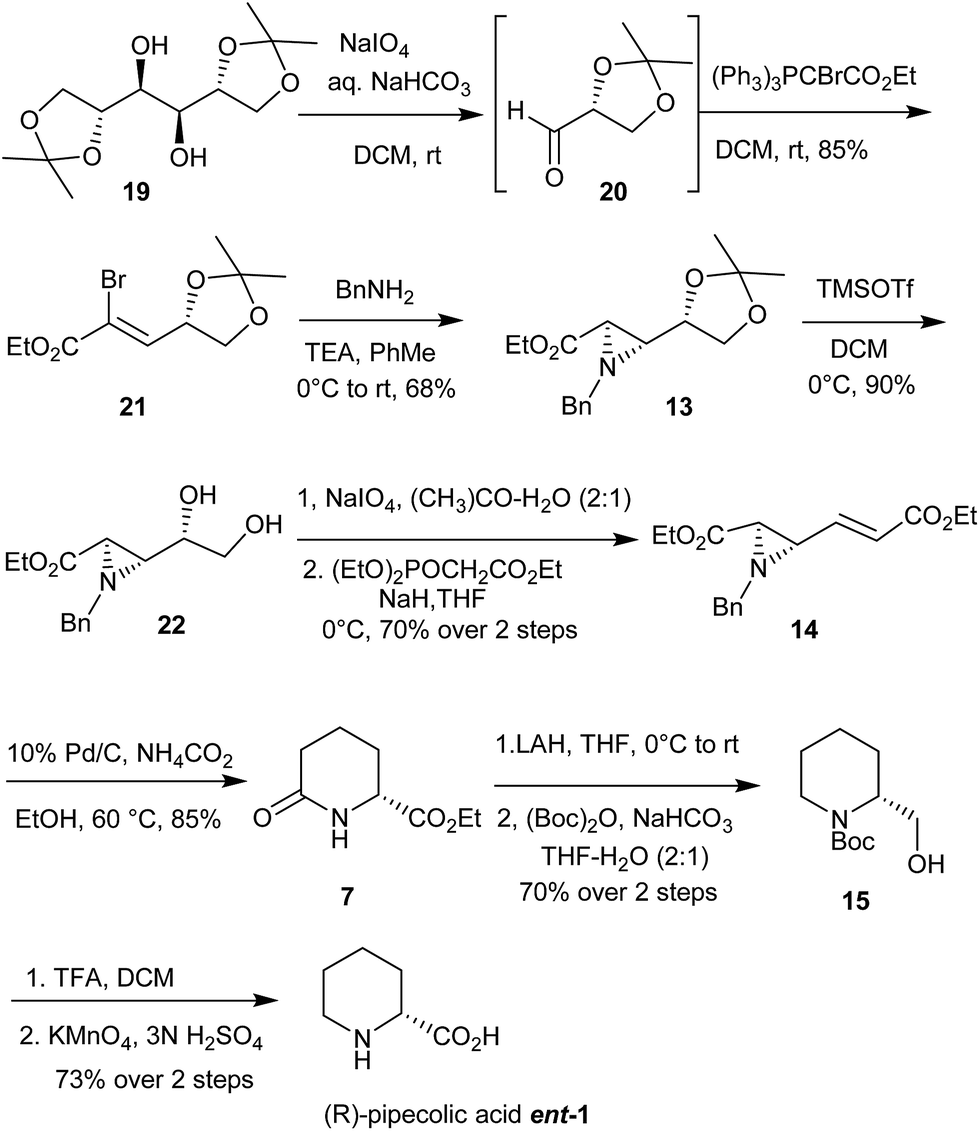
Our endeavor towards formal synthesis of (−)-swainsonine 4 and (+)-1,2-di-epi-swainsonine 5 started with aziridine-2-carboxylate 13, whose acetonide moiety was kept intact as a masked aldehyde while ester group was propagated to give trans-aziridine-α,β-unsaturated ester 17. Thus, trans-aziridne-2-carboxylate 13 on reaction with DIBAL-H (1.2 eq.) yielded aldehyde which was used as such for the next step, Horner–Wadsworth–Emmons reaction on the resultant aldehyde furnished α,β-unsaturated trans aziridine ester 17 in 75% yield over two steps. Following the aziridine ring opening reaction under acidic conditions, compound 17 gave amino alcohol 27 (Scheme 5). Hydroxyl functionality of this amino-alcohol 27 was protected as its TBS ether 28 in 90% yield. Compound 28 on palladium mediated hydrogenation/hydrogenolysis afforded lactam 29 in 92% yield. Allylation of lactam 29 was carried out using allyl bromide and NaH in DMF as the solvent to give N-allylated lactam 30 in 85% yields. Lactam 30 was exposed to 80% aqueous acetic acid at 80 °C to furnish diol 31 by selective deprotection of terminal acetonide functionality in presence of secondary –OTBS group in 75% yield.29 After failing to convert 1,2-diol functionality of the compound 31 into corresponding alkene in one step using PPh3/I2/imidazole,30 it was cleaved using NaIO4 in acetone–water to furnish the crude aldehyde which, without purification, was subjected to 2-carbon Wittig homologation in dichloromethane to yield α,β-unsaturated ester 32 in 75% yield over two steps. Finally, performing the ring closing metathesis reaction on compound 32 using Grubbs' 2nd generation catalyst31 in refluxing anhydrous dichloromethane gave access to key intermediate viz. bicyclic lactam 18 having requisite indolizidine skeleton of swainsonine. Enantiomer of 18 and its conversion to ent-4 and ent-5 is well documented in the literature. The spectral data of 18 were in good agreement with the reported one except for the sign of optical rotation (Scheme 5).
 | ||
| Scheme 5 Synthesis of (−)-swainsonine 4 and (+)-1,2-di-epi-swainsonine 5. | ||
Additionally, aziridine ester 17 when subjected to transfer hydrogenation conditions yielded lactam 33 in excellent yield. Lactam 33 is key intermediate towards the syntheses of β-(+)-conhydrine and its analogues (Scheme 6).32
 | ||
| Scheme 6 Synthesis of key intermediate 33. | ||
Conclusions
In conclusion, an efficient enantioselective total synthesis (R)-pipecolic acid ent-1, (R)-ethyl-6-oxopipecolate 7 and trans (2R,3R)-3-hydroxypipecolic acid 3. A concise formal synthesis of (−)-swainsonine 4, (+)-1,2-di-epi-swainsonine 5 and (−)-lentiginosine 6 have also been achieved from trans-aziridine-2-carboxylate 13 as the common chiral synthon. The notable features of these syntheses are regio and stereoselective Wittig olefination, ring closing metathesis, reductive cyclisation and regio and stereoselective aziridine ring opening as key chemical transformations.Syntheses are operationally simple and practical in terms of overall yield. The trans aziridine ester synthon was found to be a versatile highly efficient for the synthesis of various piperidine skeletons.
Experimental
General information
All reagents and solvents were used as received from the manufacturer. HRMS (ESI) were recorded on ORBITRAP mass analyzer (Thermo Scientific, Q Exactive). Mass spectra were measured with ESI ionization in MSQ LCMS mass spectrometer. IR spectra were recorded on a Perkin-Elmer Infrared Spectrophotometer Model 68B or on a Perkin-Elmer 1615 FT Infrared spectrophotometer. Melting points of solids were measured in Buchi melting point apparatus and are uncorrected. Optical rotation values were recorded on P-2000 polarimeter at 589 nm. 1H (200 and 400 MHz) and 13C (50 and 100 MHz) NMR spectra were recorded on Bruker and Bruker Advance 400 spectrometers, using a 1:1 mixture of CDCl3 and CCl4 as solvent. The chemical shifts (δ ppm) and coupling constants (Hz) are reported in the standard fashion with reference to chloroform, δ 7.27 (for 1H) or the central line (77.0 ppm) of CDCl3 (for 13C). In the 13C NMR spectra, the nature of the carbons (C, CH, CH2, or CH3) was determined by recording the DEPT-135 spectra. The following abbreviations were used to explain the multiplicities: br = broad, s = singlet, d = doublet, t = triplet, q = quartet. The reaction progress was monitored by the TLC analysis using thin layer plates precoated with silica gel 60 F254 (Merck) and visualized by fluorescence quenching or iodine or by charring after treatment with ethanolic solution of ninhydrin or anisaldehyde. Merck's flash silica gel (230–400 mesh) was used for column chromatography.
Experimental
:5) to give bromoester 21 (E/Z = 7:93). Rf: 0.5 (pet. ether–ethyl acetate, 9:1); yield: 10.5 g, 84% over two steps; IR (CHCl3, cm−1): νmax 2980, 1720, 1620; 1H NMR (200 MHz, CDCl3 + CCl4): δ 1.36 (t, J = 8.0 Hz, 3H), 1.41 (s, 3H), 1.46 (s, 3H), 3.70 (dd, J = 6.6 & 8.3 Hz, 1H), 4.27 (q, J = 8.0 Hz, 3H), 4.95 (dd, J = 6.7 & 13.3 Hz, 1H), 7.36 (d, J = 6.6 Hz, 1H); 13C NMR (125 MHz, CDCl3 + CCl4): δ 14.1, 25.5, 26.4, 62.6, 68.0, 75.5, 110.2, 116.7, 144.0, 161.4. MS (ESI): m/z: 279 (M + H)+.:1 which were separated using flash chromatography (pet. ether–ethyl acetate, 9:1). Yield: 75%; for 13-yield: 68%; for 33-yield: 7%.:2); IR (CHCl3, cm−1): νmax 2984, 1728, 1599, 1107. [α]25D +52.41 (c 1, CHCl3), {lit.1 [α]25D +52.8 (c 1, CHCl3)}.1H NMR (200 MHz, CDCl3 + CCl4): δ 1.19 (t, J = 8 Hz, 3H), 1.34 (s, 3H), 1.42 (s, 3H), 2.48 (t, J = 2.4 Hz, 1H), 2.63 (d, J = 2.4 Hz, 1H), 3.63–3.68 (m, 1H), 3.86–3.97 (m, 3H), 4.07–4.17 (m, 3H), 7.27–7.32 (m, 5H); 13C NMR (50 MHz, CDCl3 + CCl4): δ 14.0, 25.5, 26.6, 37.2, 47.4, 54.8, 60.1, 66.4, 75.9, 109.5, 126.9, 128.1, 138.8, 168.5; MS (ESI): m/z: 306.71 [M + H]+; HRMS: calculated for C17H24NO4-306.1700, found-306.1694.:2); IR (CHCl3, cm−1): νmax 2986, 1728, 1600, 1107; [α]25D −9.7 (c 1, CHCl3), {lit.1 [α]25D −9.9 (c 1, CHCl3)}; 1H NMR (200 MHz, CDCl3 + CCl4): δ 1.18 (t, 3H), 1.27 (s, 3H), 1.37 (s, 3H), 2.08 (t, J = 6.7 Hz, 1H), 2.23 (d, J = 6.7 Hz, 1H), 3.42 (d, J = 13.0 Hz, 1H), 3.66 (dd, J = 6.0 & 8.0 Hz, 1H), 3.89–3.97 (m, 2H), 4.11–4.22 (m, 3H), 7.27–7.35 (m, 5H); 13C NMR (50 MHz, CDCl3 + CCl4): δ 14.1, 25.3, 26.8, 40.4, 47.8, 61.0, 63.2, 66.9, 75.2, 109.6, 127.2, 127.9, 128.2, 137.2, 168.9; MS (ESI): m/z: 306.18 [M + H]+; HRMS: calculated for C17H24NO4-306.1700, found-306.1698.:3) of the residue provided the pure acetonide-cleaved product 22 as a thick liquid (0.127 g). Rf: 0.4 (pet. ether–ethyl acetate, 1:1); yield: 90%; [α]25D +20.22 (c 2.1, CHCl3), {lit.35 [α]25D +19.6 (c 0.56, CHCl3)}; IR (CHCl3, cm−1): νmax 3588, 3369, 2927, 1727, 1603, 1454, 1371, 1193; 1H NMR (200 MHz, CDCl3 + CCl4): δ 1.23 (t, J = 7.2 Hz, 3H), 2.52 (t, J = 2.9 Hz, 1H), 2.75 (d, J = 2.9 Hz, 1H), 3.26–3.32 (m, 1H), 3.44–3.50 (m, 1H), 3.61 (br s, 1H), 3.96 (s, 2H), 4.15 (q, J = 7.2 Hz, 2H), 7.27–7.31 (m, 5H); 13C NMR (50 MHz, CDCl3 + CCl4): δ 14.1, 37.4, 46.4, 54.4, 61.3, 65.2, 69.1, 127.5, 128.5, 128.6, 138.4, 168.5; MS (ESI): m/z: 266.13 (M + H) +, 288.10 (M + Na)+; HRMS: calculated for C14H20O4N-266.1387, found-266.1385.:1) at 0 °C, treated with sodium metaperiodate (0.203 g, 0.95 mmol) and stirred at 15 °C for 15 min. The reaction was quenched using ethylene glycol (0.01 mL), extracted with CH2Cl2 (3 × 15 mL), washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated to afford crude aldehyde which was used as such for next reaction. To a stirred solution of NaH (0.038 g, 1.58 mmol, prewashed with n-hexane) dissolved in THF (2 mL), was added triethyl phosphonoacetate (0.31 mL, 1.58 mmol) slowly at 0 °C and stirred for 10 minutes. The aldehyde from above reaction dissolved in dry THF (3 mL) was added and stirring continued for another 2 h at same temperature until completion of reaction. The reaction was quenched with saturated aqueous NH4Cl (10 mL) and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were then washed with brine (10 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo. Purification on flash column chromatography (pet. ether–ethyl acetate, 9:1) furnished compound 14 (0.168 g) as thick colorless oil. Rf: 0.5 (pet. ether–ethyl acetate, 4:1); yield: 0.168 g, 70%; [α]25D −36 (c 1, CHCl3); IR (CHCl3, cm−1): νmax 2926, 2850, 1720, 1651, 1456, 1368, 1265, 1180, 1030; 1H NMR (200 MHz, CDCl3 + CCl4): δ 1.22–1.32 (m, 6H), 2.55–2.73 (m, 1H), 2.46–3.17 (m, 1H), 3.84–4.20 (m, 8H), 6.05–6.25 (m, 1H), 6.60–6.89 (m, 1H); 13C NMR (50 MHz, CDCl3 + CCl4): δ 14.0, 14.1, 42.9, 45.9, 54.4, 60.2, 61.1, 123.3, 127.6, 127.9, 128.2, 138.4, 145.3, 165.4, 167.8; doubling of peaks in 1H and 13C is attributed to invertomerism; MS (ESI): m/z: 303.28 (M)+, 326.21 (M + Na)+; HRMS: calculated for C17H21NO4Na-326.1363, found-326.1358.:90) to yield 0.071 g of amide-ester 7 as colourless liquid. Rf: 0.3 (ethyl acetate); yield: 85%; [α]25D +13.4 (c 1.4, CHCl3) {lit.36 for ent-7, [α]25D −13.7 (c 0.3, CHCl3)}; IR (CHCl3, cm−1): νmax 2958, 1739, 1666, 1468, 1198; 1H NMR (200 MHz, CDCl3 + CCl4): δ 1.30 (t, J = 7.3 Hz, 3H), 1.78–1.98 (m, 3H), 2.20–2.22 (m, 1H), 2.36–2.47 (m, 2H), 4.1 (dd, J = 5.5 & 7.0 Hz, 1H), 4.24 (qd, J = 1.2 & 7.3 Hz, 2H), 6.65 (br s, 1H); 13C NMR (100 MHz, CDCl3 + CCl4): δ 14.1, 19.2, 25.2, 30.7, 54.7, 61.9, 170.7, 171.9; MS (ESI): m/z: 194.08 (M + Na)+; HRMS: calculated for C8H14O3N-172.0968, found-172.0966.:1) was added solid NaHCO3 (0.2 g, 2.34 mmol) and (Boc)2O (0.536 mL, 2.34 mmol) and then the mixture was vigorously stirred at room temperature for 6 h. The reaction mixture was extracted with ethyl acetate (3 × 10 mL), washed with brine, dried over anhydrous Na2SO4, filtered, concentrated in vacuo and purified by column chromatography (pet. ether–ethyl acetate, 8:2) to afford 15 as a white solid. Rf: 0.5 (pet. ether–ethyl acetate, 8:2); yield: 0.176 g, 70% over two steps; MP: 81–84 °C, lit.37 81–84 °C; [α]25D +38.5 (c 1, CHCl3) {For ent-15 lit.5 [α]25D −40.5 (c 1, CHCl3)}; IR (CHCl3, cm−1): νmax 3443, 2940, 2890, 1655, 1280; 1H NMR (200 MHz, CDCl3 + CCl4): δ 1.46 (s, 9H), 1.60–1.65 (m, 6H), 2.11 (br s, 1H), 2.87 (t, J = 13.0 Hz, 1H), 3.59 (dd, J = 5.9 & 11.0 Hz, 1H), 3.79 (dd, J = 9.0 & 11.0 Hz, 1H), 3.93 (br d, J = 13.5 Hz, 1H), 4.25–4.29 (m, 1H); 13C NMR (100 MHz, CDCl3 + CCl4): δ 19.3, 24.8, 25.1, 28.3, 39.7, 52.0, 60.6, 79.4, 155.8; MS (ESI): m/z: 238 (M + Na)+; HRMS: calculated for C11H22O3N-216.1954, found-216.1600; HPLC detail for racemic hydroxy compound (15): HPLC Kromacil 5-Amycoat column (250 × 4.6 mm). Isopropanol–n-hexane = 4:96; flow rate 0.5 mL min−1, λ = 210 nm retention time (min): rt1 = 22.18; rt2 = 24.05 (1:1). Enantiomerically pure hydroxy compound (15) HPLC Kromacil 5-Amycoat column (250 × 4.6 mm) isopropanol–n-hexane = 4:96; flow rate 0.5 mL min−1, ë = 210 nm retention time (min): rt1 = 22.07 (major); rt2 = 24.02 (>97% ee).:1:1%); MP: 271–273 °C; lit.38 271–274 °C; [α]25D +24.9 (c 1.15, H2O) {lit.5 [α]25D +25.8 (c 1, H2O)}; 1H NMR (400 MHz, D2O): δ 1.46–1.64 (m, 3H), 1.73–1.80 (m, 2H), 2.14–2.18 (m, 1H), 2.87–2.94 (m, 1H), 3.31–3.54 (m, 1H), 3.78 (dd, J = 8.0 Hz & 10.0 Hz, 1H); 13C NMR (100 MHz, D2O): δ 21.5, 21.6, 26.0, 44.0, 57.2, 172.2; MS (ESI): m/z: 152.28 (M + Na)+.:1, 20 mL) was added TFA (0.45 mL, 7.79 mmol) drop wise at 0 °C. The reaction mixture was slowly warmed to room temperature and stirred until complete disappearance of starting material (∼5–6 h). Reaction was quenched by excess NaHCO3, water (10 mL) was added and organic mass was extracted with ethyl acetate (3 × 15 mL). Combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure followed by column chromatographic purification using ethyl acetate–pet ether (15:85) to yield 0.95 g of amino-alcohol 16 as thick liquid. Rf: 0.5 (pet ether–ethyl acetate, 7:3); yield: 76% over two steps; [α]25D +20 (c 0.5, CHCl3); IR (CHCl3, cm−1): νmax 3554, 3359, 2980, 1720, 1620; 1H NMR (200 MHz, CDCl3 + CCl4): 1.26–1.34 (m, 6H), 3.53 (d, J = 5 Hz, 1H), 3.68 (d, J = 13 Hz, 1H), 3.94 (d, J = 13 Hz, 1H), 4.13–4.28 (m, 4H), 4.52–4.56 (m, 1H), 6.08 (dd, J = 2 & 15.5 Hz, 1H), 6.75 (dd, J = 4.0 & 15.5 Hz, 1H), 7.27–7.30 (m, 5H); 13C NMR (50 MHz, CDCl3 + CCl4): 14.2, 52.6, 60.3, 61.3, 64.1, 70.1, 122.7, 127.5, 128.2, 128.4, 138.9, 145.2, 165.7, 171.6; MS (ESI): m/z: 344.18 (M + Na)+; HRMS: calculated for C17H24O5N-322.1649, found-322.1640.:95) to yield 0.8 g of hydroxy amino TBS ether 23 as thick colorless liquid. Rf: 0.5 (pet. ether–ethyl acetate, 8:2); yield: 85% over two steps. [α]25D −7.69 (c 1, CHCl3); IR (CHCl3, cm−1): νmax 2980, 1720, 1620; 1H NMR (500 MHz, CDCl3 + CCl4): 0.01 (s, 3H), 0.03 (s, 3H), 0.87 (s, 9H), 1.24–1.31 (m, 6H), 2.17 (br s, 1H), 3.28 (d, J = 5.5 Hz, 1H), 3.65 (d, J = 13 Hz, 1H), 3.84 (d, J = 13.0 Hz, 1H), 4.12–4.20 (m, 5H), 4.47–4.48 (m, 1H), 5.95 (dd, J = 1.5 & 15.5 Hz, 1H), 6.95 (dd, J = 5.2 & 15.5 Hz, 1H), 7.21–7.28 (m, 5H); 13C NMR (125 MHz, CDCl3 + CCl4): −4.6, −4.4, 14.3, 18.1, 25.7, 52.2, 60.3, 60.7, 65.7, 73.7, 121.7, 127.1, 128.2, 128.3, 139.4, 147.5, 166.0, 172.0; MS (ESI): m/z: 436.68 (M + H)+; HRMS: calculated for C23H38ON5Si-436.2514, found-436.2505.:3) to provide amide 24 (0.52 g) as a colorless thick oil. Rf: 0.4 (pet. ether–ethyl acetate, 8:2); yield: 85%; [α]25D −26 (c 1.5, CHCl3); IR (CHCl3, cm−1): νmax 3399, 2955, 2857, 1732, 1643, 1215; 1H NMR (200 MHz, CDCl3): δ 0.12 (s, 6H), 0.90 (s, 9H), 1.30 (t, J = 7.2 Hz, 3H), 1.68 (br s, 1H), 1.79–1.88 (m, 2H), 2.26–2.40 (m, 1H), 2.54–2.72 (m, 1H), 3.99–4.02 (m, 1H), 4.23 (q, J = 7.2 Hz, 2H), 4.35–4.39 (m, 1H), 5.96 (br s, 1H); 13C-NMR (50 MHz, CDCl3): δ −5.1, −4.9, 14.1, 17.9, 25.6, 26.4, 26.5, 61.8, 62.3, 65.4, 170.1, 171.4. MS (ESI): m/z: 302.2 (M + H)+; HRMS: calculated for C14H28O4NSi-302.1782, found-302.1777.:30, EtOAc–pet ether) to furnish amine 25 (0.147 g, 78%) as a colorless dense liquid. Rf: 0.5 (pet. ether–ethyl acetate, 2:8); yield: 78%; [α]25D −27 (c 1.0, CHCl3); IR (CHCl3, cm−1): νmax 3436, 3020, 2931, 2400, 1731, 1215 cm−1; 1H NMR (200 MHz, CDCl3 + CCl4): δ 0.00 (s, 3H), 0.06 (s, 3H), 0.86 (s, 9H), 1.36 (t, J = 7.3 Hz, 3H), 1.41–1.51 (m, 2H), 1.58–1.68 (m, 2H), 1.84–1.87 (m, 1H), 2.01–2.05 (m, 1H), 2.53–2.64 (m, 1H), 3.11 (dd, J = 1.1 & 10.1 Hz, 1H), 3.32 (d, J = 13.5 Hz, 1H), 3.78 (dt, J = 5.4 & 10.5 Hz, 1H), 3.98 (m, 1H), 4.10–4.18 (m, 1H), 4.31–4.39 (m, 1H); 13C-NMR (100 MHz, CDCl3 + CCl4): δ −5.3, −4.2, 13.9, 17.8, 23.1, 25.5, 32.2, 52.2, 61.8, 70.2, 70.5, 170.8; MS (ESI): m/z: 288.23 (M + H)+, 310.14 (M + Na)+; HRMS: calculated for C14H30O3NSi-288.1989, found-288.1979.:1:1%); yield: 91%; MP: 238–243 °C (dec.), lit.39 230–238 °C; [α]25D −13.8 (c 1.0, aq. HCl 10%), {lit.7 [α]25D −14 (c 0.5, aq. HC1 10%)}; IR (CHCl3, cm−1): νmax 3287, 2920, 1625, 1405 cm−1; 1H NMR (400 MHz, D2O): δ 1.64–1.80 (m, 2H), 2.02–2.08 (m, 2H), 2.22 (s, 1H), 3.07–3.12 (m, 1H), 3.40–3.36 (m, 1H), 3.83 (d, J = 7.8 Hz, 1H), 4.17–4.13 (m, 1H); 13C-NMR (100 MHz, D2O): δ 18.5, 28.7, 42.5, 60.8, 65.5, 170.0; MS (ESI): m/z: 146 (M + H)+.:90) to afford diol 26 (138 mg) as a white crystalline solid. Rf: 0.5 (pet. ether–ethyl acetate, 2:8); yield: 75%; MP: 126–128 °C, lit.8 124–126 °C; [α]25D +27 (c 1.0, MeOH), {lit.40 [α]25D +29.8 (c 0.99, MeOH)}; IR (CHCl3, cm−1): νmax 3448, 3025, 2945, 1674, 1215, 1120, 838 cm−1; 1H NMR (200 MHz, CDCl3 + CCl4 + DMSO-d6): δ 1.15–1.29 (m, 1H), 1.39 (s, 9H), 1.61–1.82 (m, 3H), 2.69–2.82 (m, 1H), 3.45–3.61 (m, 2H), 3.89–3.92 (m, 2H), 4.08–4.16 (m, 1H); 13C (125 MHz, CDCl3 + CCl4 + DMSO-d6): δ 18.8, 26.3, 28.0, 39.6, 59.1, 59.8, 63.8, 79.1, 155.9; MS (ESI): m/z: 232 (M + H)+, 254 (M + Na)+; HRMS: calculated for C11H21NNaO4-254.1368, found-254.1369. HPLC detail for racemic dihydroxy compound (26) HPLC chiracel OJ-H column (250 × 4.6 mm). Isopropanol–pet ether = 5:95 flow rate 0.5 mL min−1, λ = 210 nm retention time (min): rt1 = 13.39; rt2 = 14.98 (1:1). Enantiomerically pure dihydroxy compound (26) HPLC chiracel OJ-H column (250 × 4.6 mm) isopropanol–pet ether = 5:95 flow rate 0.5 mL min−1, λ = 210 nm retention time (min): rt1 = 13.18 (major); rt2 = 15.05 (>97% ee).:9) furnished compound 17 (0.75 g) as thick colorless oil. Rf: 0.5 (pet ether–ethyl acetate, 8:2); yield: 75%, over two steps; IR (CHCl3, cm−1): νmax 2984, 2932, 1716, 1644, 1370, 1265; [α]25D −34 (c 1, CHCl3); 1H NMR (200 MHz, CDCl3 + CCl4): δ 1.30 (t, J = 7.0 Hz, 3H), 1.33 (s, 3H), 1.40 (s, 3H), 2.12 (dd, J = 2.6 & 4.9 Hz, 1H), 2.72 (dd, J = 2.4 & 9.9 Hz, 1H), 3.61 (dd, J = 5.5 & 7.9 Hz, 1H), 3.72–3.85 (m, 2H), 3.91–4.09 (m, 2H), 4.20 (q, J = 7.0 Hz, 2H), 6.13 (d, J = 15.2 Hz, 1H), 6.89 (dd, J = 9.9 & 15.2 Hz, 1H), 7.26–7.35 (m, 5H); 13C NMR (100 MHz, CDCl3 + CCl4): δ 14.2, 25.5, 26.7, 40.1, 49.6, 57.0, 60.3, 66.24, 76.1, 109.5, 125.1, 127.1, 127.9, 128.2, 138.6, 142.9, 165.3; MS (ESI): m/z: 354.15 [M + Na]+; HRMS: calculated for C19H26O4N-332.1862, found-332.1858.:1, 25 mL) was added TFA (0.64 mL, 8.4 mmol) dropwise at 0 °C. The reaction mixture was slowly warmed to room temperature and stirred until complete disappearance of starting material (∼5–6 h). Reaction was quenched by addition of excess NaHCO3, water (10 mL) was added and organic mass was extracted with ethyl acetate (3 × 20 mL). Combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure followed by column chromatographic purification using ethyl acetate–pet. ether (15:85) to yield 1.11 g of amino-alcohol 27 as thick liquid. Rf: 0.5 (pet. ether–ethyl acetate, 7:3); yield: 80%; IR (CHCl3, cm−1): νmax 3453, 2985, 1717, 1656, 1455, 1370, 1263, 1175; [α]25D −50 (c 1.8, CHCl3); 1H NMR (200 MHz, CDCl3 + CCl4): δ 1.29 (t, J = 7.0 Hz, 3H), 1.32 (s, 3H), 1.39 (s, 3H), 2.74 (dd, J = 3.6 & 5.4 Hz, 1H), 3.77–4.03 (m, 4H), 4.1–4.26 (m, 3H), 4.55 (dd, J = 3.6 & 5.4 Hz, 1H), 6.2 (dd, J = 2 & 15.6 Hz, 1H), 6.9 (dd, J = 3.7 & 15.6 Hz, 1H), 7.26–7.34 (m, 5H); 13C NMR (50 MHz, CDCl3): δ 14.1, 25.1, 26.3, 51.0, 60.3, 61.3, 67.3, 69.0, 74.9, 109.0, 121.3, 127.1, 128.1, 128.4, 139.5, 147.0, 166.1; MS (ESI): m/z: 372.14 [M + Na]+. HRMS: calculated for C19H28O5N-350.1967, found 350.1962.:95) to yield 1.18 g of –OTBS protected amino-alcohol 28 as thick colourless liquid. Rf: 0.5 (pet. ether–ethyl acetate, 8:2); yield: 90%; IR (CHCl3, cm−1): νmax 2984, 2931, 1721, 1657, 1472, 1369, 1260, 1160, 1059. [α]25D +11.11 (c 2.7, CHCl3); 1HNMR (200 MHz, CDCl3 + CCl4): δ 0.04 (s, 3H), 0.09 (s, 3H), 0.91 (s, 9H), 1.31 (t, 3H), 1.35 (s, 3H), 1.40 (s, 3H), 2.72 (br s, 1H), 3.70 (t, J = 7.7 Hz, 1H), 3.84–4.04 (m, 1H), 4.22 (q, 2H), 4.29–4.46 (m, 2H), 6.08 (dd, J = 1.4 & 15.6 Hz, 1H), 7.1 (dd, J = 5.2 & 15.6 Hz, 1H), 7.25–7.36 (m, 5H); 13C NMR (50 MHz, CDCl3): δ −4.9, −4.5, 14.1, 18.1, 25.2, 25.8, 26.8, 53.1, 60.3, 63.7, 66.9, 73.3, 75.5, 108.8, 121.4, 126.9, 128.2, 149.0, 166.2; MS (ESI): m/z: 486.27 [M + Na]+; HRMS: calculated for C25H42O5NSi-464.2832, found-464.2827.:3) to afford amide 29 (0.59 g) as a colorless thick liquid. Rf: 0.5 (pet. ether–ethyl acetate, 1:1); yield: 92%; IR (CHCl3, cm−1): νmax 3408, 2927, 1670, 1457, 1380, 1216; [α]25D −22.9 (c 1.15, CHCl3). 1HNMR (400 MHz, CDCl3 + CCl4): δ 0.1 (s, 3H), 0.11 (s, 3H), 0.91 (s, 9H), 1.34 (s, 3H), 1.41 (s, 3H), 1.78–1.87 (m, 1H), 1.94–2.01 (m, 1H), 2.29–2.38 (m, 1H), 2.47–2.85 (m, 1H), 3.20 (t, J = 7 Hz, 1H), 3.72–3.77 (m, 1H), 3.84 (dd, J = 5 & 8 Hz, 1H), 4.00–4.1 (m, 2H), 6.02 (br s, 1H); 13C NMR (100 MHz, CDCl3 + CCl4): δ −4.5, −4.1, 17.9, 25.2, 25.8, 26.6, 28.5, 29.1, 61.7, 67.2, 68.1, 76.3, 109.3, 170.6; MS (ESI): m/z: 352.18 [M + Na]+; HRMS: calculated for C16H32O4NSi-330.2101, found-330.2095.:3) to afford the allylated product 30 as colorless liquid. Rf: 0.5 (pet. ether–ethyl acetate, 2:1); yield: 0.357 g, 85%; IR (CHCl3, cm−1): νmax 2986, 1630, 1420, 1107. [α]25D −83.4 (c 1, CHCl3); 1H NMR (200 MHz, CDCl3 + CCl4): δ 0.07 (s, 3H), 0.08 (s, 3H), 0.88 (s, 9H), 1.35 (s, 3H), 1.43 (s, 3H), 1.88–1.92 (m, 4H), 2.33–2.44 (m, 1H)), 2.53–2.67 (m, 1H), 3.33–3.37 (m, 1H), 3.54–3.75 (m, 3H), 4.03–4.05 (m, 2H), 4.9 (m, 1H), 5.11–5.24 (m, 2H), 5.61–5.81 (m, 1H); 13C NMR (50 MHz, CDCl3 + CCl4): δ −4.9, 17.8, 25.4, 25.5, 25.6, 26.3, 26.5, 48.4, 64.4, 65.4, 66.7, 78.5, 109.6, 117.1, 133.4, 168.8; MS (ESI): m/z: 356.41 [M + H]+.:9). Pure terminal diol 31 (0.14 g) was obtained as a thick gummy liquid. Rf: 0.4 (ethyl acetate); yield: 75%; IR (CHCl3, cm−1): νmax 3554, 3340, 2986, 1627, 1423, 1107; [α]25D −34.9 (c 1, CHCl3); 1HNMR (200 MHz, CDCl3 + CCl4): δ 0.06 (s, 3H), 0.07 (s, 3H), 0.88 (s, 9H), 1.99–2.11 (m, 2H), 2.14–2.33 (m, 1H), 2.53–2.63 (m, 1H), 3.35–3.37 (m, 1H), 3.54–3.69 (m, 4H), 3.94 (s, 1H), 4.71–4.77 (m, 2H), 5.26–5.58 (m, 2H), 5.72–5.88 (m, 1H); 13C NMR (100 MHz, CDCl3 + CCl4): −4.8, −4.7, 18.0, 25.3, 25.8, 26.9, 50.3, 64.1, 64.7, 66.1, 73.6, 117.5, 132.8, 171.0; MS (ESI): m/z: 352.23 [M + Na]+.:1) at 0 °C, treated with sodium metaperiodate (0.2 g, 0.9 mmol) and stirred at 15 °C for 15 min. The reaction was quenched using ethylene glycol (0.01 mL), extracted with CH2Cl2 (3 × 10 mL), washed with brine, dried over anhydrous Na2SO4 and filtered. The combined organics were concentrated under reduced pressure to afford crude aldehyde which was used as such for next reaction.To a solution of aldehyde from above reaction in CH2Cl2 (15 mL) was added (carboethoxymethylene) triphenylphosphorane (0.4 g, 1.2 mmol) and the reaction mixture was stirred for 6 h. Solvent was evaporated and the reaction mixture was adsorbed on silica. Purification by column chromatography (pet. ether–ethyl acetate, 8:2) gave 32 as a thick liquid (0.167 g). Rf: 0.5 (pet. ether–ethyl acetate, 1:1); yield: 75%; IR (CHCl3, cm−1): νmax 2986, 1723, 1656, 1630, 1107; [α]25D −45 (c 1, CHCl3) 1HNMR (200 MHz, CDCl3 + CCl4): δ 0.06 (s, 3H), 0.08 (s, 3H), 0.87 (s, 9H), 1.30 (t, J = 7 Hz, 3H), 1.73–1.75 (m, 1H), 1.86–1.93 (m, 1H)), 2.35 (m, 1H), 2.59–2.68 (m, 1H), 2.99 (dd, J = 7 & 16 Hz, 1H), 3.99 (m, 1H), 4.19 (q, J = 7 Hz, 2H), 5.84 (dt, J = 2 & 16 Hz, 1H), 5.11–5.18 (m, 1H), 5.62–5.72 (m, 1H), 5.88 (dd, J = 1 & 16 Hz, 1H), 6.73 (dd, J = 6 & 16 Hz, 1H); 13C NMR (50 MHz, CDCl3 + CCl4): δ −4.8, 14.2, 24.8, 25.6, 26.7, 47.3, 60.7, 64.3, 67.0, 117.3, 123.9, 132.3, 144.5, 165.4, 169.1; MS (ESI) m/z: 390.12 [M + Na]+; HRMS: calculated for C19H34O4NSi-368.2252, found-368.2247.
:1) to provide the ring closed product 18 (0.044 g, 80%) as a colorless sticky liquid. Rf: 0.3 (pet. ether–ethyl acetate, 1:1); yield: 80%; IR (CHCl3, cm−1): νmax 1640, 1620; [α]25D + 53 (c 1, CHCl3); lit.41 {for ent-[α]25D −53.73 (c 1.10, CHCl3)}; 1HNMR (400 MHz, CDCl3 + CCl4): δ 0.08 (s, 6H), 0.90 (s, 9H), 1.79–1.81 (m, 1H), 2.02–2.03 (m, 1H), 2.39–2.46 (m, 1H), 2.60–2.62 (m, 1H), 3.53–3.55 (m, 1H), 4.02–4.06 (m, 1H), 4.15–4.16 (m, 1H), 4.45–4.50 (m, 1H), 5.92–5.94 (m, 2H); 13C NMR (100 MHz, CDCl3 + CCl4): δ −4.6, −4.1, 18.0, 25.7, 29.7, 30.2, 53.3, 69.1, 71.1, 126.8, 128.5, 168.2. MS (ESI): m/z: 268.02 [M + H]+. HRMS: calculated for C14H26NO2Si-268.1733; found-268.1741.:8) to afford 33 as a thick yellowish liquid. Rf: 0.4 (ethyl acetate); yield: 0.37 g, 95%; [α]25D −17.5 (c 1.1, CHCl3); {lit.42 [α]25D −14.4, (c 0.5, CHCl3)}; IR (CHCl3, cm−1): νmax 3402, 2985, 2936, 1664, 1457, 1371, 1072; 1H NMR (400 MHz, CDCl3 + CCl4): δ 1.20–1.28 (m, 1H), 1.33 (s, 3H), 1.40 (s, 3H), 1.63–1.83 (m, 2H), 1.85–2.01 (m, 1H), 2.17–2.49 (m, 2H), 3.31 (td, J = 5.4 & 8.7 Hz, 1H), 3.66 (dd, J = 5.4 & 8.2 Hz, 1H), 3.86–3.88 (m, 1H), 4.03 (dd, J = 6.0 & 8.2 Hz, 1H), 6.21 (br s, 1H); 13C NMR (100 MHz, CDCl3 + CCl4): δ 19.7, 24.8, 25.3, 26.8, 31.3, 56.2, 66.2, 79.1, 109.79, 171.2; MS (ESI): m/z: 200.11 (M + H)+, 222.10 (M + Na)+; HRMS: calculated for C7H18NO3-200.1281, found-200.1277.Acknowledgements
LBK, KPP, PNC and SAK thank CSIR, New Delhi, India for fellowship. We also thank Dr U. R. Kalkote, Dr H. B. Borate, Dr N. B. Dumare and Mr D. B. Kalbhor for helpful discussions. Authors thank CSIR, New Delhi for financial support as XII year plan programme under title ORIGIN (CSC-0108) and ACT (CSC-0301).Notes and references
- Review articles: (a) M. He, J. Ind. Microbiol. Biotechnol., 2006, 33, 401 CrossRef CAS PubMed; (b) H. P. Broquist, Annu. Rev. Nutr., 1991, 11, 435 CrossRef CAS PubMed; (c) V. Vranova, L. Lojkova, K. Rejsek and P. Formanek, Chirality, 2013, 25, 823 CrossRef CAS PubMed.
- (a) D. C. N. Swindells, P. S. White and J. A. Findlay, Can. J. Chem., 1978, 56, 2491 CrossRef CAS; (b) A. B. Smith III, K. J. Hale, L. M. Laakso, K. Chen and A. Riéra, Tetrahedron Lett., 1989, 30, 6963 CrossRef; (c) A. B. Smith III, S. M. Condon, J. A. McCauley, J. L. Leazer Jr, J. W. Leahy and R. E. Maleczka Jr, J. Am. Chem. Soc., 1997, 119, 962 CrossRef; (d) A. B. Smith III and C. M. Adams, Acc. Chem. Res., 2004, 37, 365 CrossRef PubMed.
- (a) H. Tanaka, A. Kuroda, H. Marusawa, H. Hatanaka, T. Kino, T. Goto, M. Hashimoto and T. Taga, J. Am. Chem. Soc., 1987, 109, 5031 CrossRef CAS; (b) D. Romo, S. D. Meyer, D. D. Johnson and S. L. Schreiber, J. Am. Chem. Soc., 1993, 115, 7906 CrossRef CAS; (c) R. E. Ireland, J. L. Gleason, L. D. Gregnas and T. K. A. Highsmith, J. Org. Chem., 1996, 61, 6856 CrossRef CAS PubMed.
- D. L. Boger, J.-H. Chen and K. W. Saionz, J. Am. Chem. Soc., 1996, 118, 1629 CrossRef CAS.
- J. B. Rangisetty, M. R. Pullagurla, R. J. J. Muthiah and C. N. Jobdevairakkam, US pat. 7683175, 2008.
- (a) T. Fujii, M. Mukaihara, H. Agematu and H. Tsunekawa, Biosci., Biotechnol., Biochem., 2002, 66, 622 CrossRef CAS; (b) S. B. Singh, D. L. Zink, J. M. Liesch, R. T. Mosley, A. W. Dombrowski, G. F. Bills, S. J. Darkin-Rattray, D. M. Schmatz and M. A. Goetz, J. Org. Chem., 2002, 67, 815 CrossRef CAS PubMed; (c) A. V. Bieliauskas and M. K. H. Pflum, Chem. Soc. Rev., 2008, 37, 1402 RSC.
- I. Pastuszak, R. J. Molyneux, L. F. James and A. D. Elbein, Biochemistry, 1990, 29, 1886 CrossRef CAS.
- M. A. Wijdeven, J. Willemsen and F. P. J. T. Rutjes, Eur. J. Org. Chem., 2010, 2831 CrossRef CAS PubMed.
- F. Ferreira, C. Greck and J. P. Genet, Bull. Soc. Chim. Fr., 1997, 134, 615 CAS.
- (a) M. J. Humphries, K. Matsumoto, S. L. White, R. J. Molyneux and K. Olden, Cancer Res., 1988, 48, 1410 CAS; (b) J. W. Dennis, Cancer Res., 1986, 46, 5131 CAS; (c) C. Galustian, S. Foulds, J. F. Dye and P. J. Guillou, Immunopharmacology, 1994, 27, 165 CrossRef CAS; (d) P. C. Das, J. D. Roberts, S. L. White and K. Olden, Oncol. Res., 1995, 7, 425 CAS; (e) H. Motohiro, N. Kunio, T. Hiroshi, H. Junji, K. Masanobu, A. Hatsuo and I. Hiroshi, Patent EP 104826, 1983 Chem. Abstr., 1984, 101, 28283x.
- (a) F. A. Kuehl Jr, C. F. Spencer and K. Folkers, J. Am. Chem. Soc., 1948, 70, 2091 CrossRef CAS; (b) S. Kobayashi, M. Ueno and R. Suzuki, Tetrahedron Lett., 1999, 40, 2175 CrossRef CAS.
- (a) G. Ratle, X. Monseur, B. C. Das, J. Yassi, Q. Khuong-Huu and R. Goutarel, Bull. Soc. Chim. Fr., 1966, 2945 CAS; (b) Q. Khuong-Huu, G. Ratle, X. Monseur and R. Goutarel, Bull. Soc. Chim. Belg., 1972, 81, 425 CrossRef PubMed; (c) Q. Khuong-Huu, G. Ratle, X. Monseur and R. Goutarel, Bull. Soc. Chim. Belg., 1972, 81, 443 CrossRef PubMed; (d) P. Bourinet and A. Quevauviller, C. R. Seances Soc. Biol. Ses Fil., 1968, 162, 1138 Search PubMed[CA 70: 95233k] (e) P. Bourinet and A. Quevauviller, Ann. Pharm. Fr., 1968, 26, 787 Search PubMed[CA 71: 29012g].
- J. D. Scott, T. N. Tippie and R. M. Williams, Tetrahedron Lett., 1998, 39, 3659 CrossRef CAS.
- (a) S.-B. Huang, J. S. Nelson and D. D. Weller, Synth. Commun., 1989, 19, 3485 CrossRef CAS PubMed; (b) A. Sadiq and N. Sewald, ARKIVOC, 2012, 5, 28 Search PubMed; (c) S. A. Hermitage and M. G. Moloney, Tetrahedron: Asymmetry, 1994, 5, 1463 CrossRef CAS; (d) T. J. Hodgkinson and M. Shipman, Synthesis, 1998, 1141 CrossRef CAS PubMed; (e) P. K. Upadhyay and P. Kumar, Synthesis, 2010, 15, 2512 Search PubMed.
- For reviews, see: (a) S. Hanessian, G. McNaughton-Smith, H.-G. Lombart and W. D. Lubell, Tetrahedron, 1997, 53, 12789 CrossRef CAS; (b) S. M. Cowell, Y. S. Lee, J. P. Cain and V. J. Hruby, Curr. Med. Chem., 2004, 11, 2785 CrossRef CAS; (c) T. D. Copeland, E. M. Wondrak, J. Toszer, M. M. Roberts and S. Oraszan, Biochem. Biophys. Res. Commun., 1990, 169, 310 CrossRef CAS; (d) M. Quibell, A. Benn, N. Flinn, T. Monk, M. Ramjee, Y. Wang and J. Watts, Bioorg. Med. Chem., 2004, 12, 5689 CrossRef CAS PubMed.
- (a) P. H.-Y. Cheoug, H. Zhang, R. Thayumanavan, F. Tanaka, K. N. Houk and C. F. Barbas III, Org. Lett., 2006, 8, 811 CrossRef PubMed; (b) C. E. Aroyan, M. M. Vasbinder and S. Miller, Org. Lett., 2005, 7, 3849 CrossRef CAS PubMed; (c) X. Liu, L. Lin and X. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef CAS PubMedand references cited therein.
- For recent syntheses of pipeclic acid: (a) A. Sadiq and N. Sewald, J. Amino Acids, 2013, 8 Search PubMed; (b) R. A. Fernandes and J. L. Nallasivam, Org. Biomol. Chem., 2012, 10, 7789 RSC; (c) T. K. Beng and R. E. Gawley, J. Am. Chem. Soc., 2010, 132, 12216 CrossRef CAS PubMed; (d) B. V. S. Reddy, D. N. Chaya, J. S. Yadav and R. Gree, Synthesis, 2012, 297 CrossRef CAS PubMedFor reviews on syntheses of pipecolic acid and derivatives, see: (e) C. Agami, F. Couty and C. Puchot-Kadouri, Synlett, 1998, 449 CrossRef CAS PubMed; (f) F. Couty, Amino Acids, 1999, 16, 297 CrossRef CAS; (g) F. P. J. T. Rutjes, L. B. Wolf and H. E. J. Schoemaker, J. Chem. Soc., Perkin Trans. 1, 2000, 4197 RSC; (h) K.-H. Park and M. J. Kurth, Tetrahedron, 2002, 58, 8629 CrossRef CAS; (i) C. Kadouri-Puchot and S. Comesse, Amino Acids, 2005, 29, 101 CrossRef CAS PubMed; (j) A. A. Cant and A. Sutheland, Synthesis, 2012, 1935 CAS; (k) S. Mohapatra, S. Bhakta, N. Baral and S. Nayak, Res. Chem. Intermed., 2014, 1–9 Search PubMedFor some recent synthesis of 3-hydroxypipecolic acid: (l) O. K. Karjalainen and A. M. P. Koskinen, Tetrahedron, 2014, 70, 2444 CrossRef CAS PubMed; (m) S. V. Pansare and E. K. Paul, Org. Biomol. Chem., 2012, 10, 2119 RSC; (n) S. Begliomini, L. Sernissi, D. Scarpi and E. G. Occhiato, Eur. J. Org. Chem., 2014, 5448 CrossRef CAS PubMedFor review on synthesis of 3-hydroxypipecolic acid: (o) A. Cochi, D. Gomez Pardo and J. Cossy, Eur. J. Org. Chem., 2013, 809 CrossRef CAS PubMed.
- For recent syntheses of lentiginosine, see: (a) S. Du-a-man, D. Soorukram, C. Kuhakarn, P. Tuchinda, V. Reutrakul and M. Pohmakotr, Eur. J. Org. Chem., 2014, 1708 CrossRef CAS PubMed; (b) S. V. Kauloorkar, V. Jha, G. Jogdand and P. Kumar, Org. Biomol. Chem., 2014, 12, 4454 RSC; (c) G.-W. Kim, T. Jin, J.-S. Kim, S.-H. Park, K.-H. Lee, S.-S. Kim, I.-S. Myeong and W.-H. Ham, Tetrahedron: Asymmetry, 2014, 25, 87 CrossRef CAS PubMed; (d) M. Lingamurthy, A. Rajender and B. V. Rao, Tetrahedron: Asymmetry, 2014, 25, 860 CrossRef CAS PubMed; (e) H. Yoon, K. S. Cho and T. Sim, Tetrahedron: Asymmetry, 2014, 25, 497 CrossRef CAS PubMed; (f) J. Zeng, Q. Zhang, H.-K. Zhang and A. Chen, RSC Adv., 2013, 3, 20298 RSC; (g) J. Shao and J.-S. Yang, J. Org. Chem., 2012, 77, 7891 CrossRef CAS PubMed; (h) S.-W. Liu, H.-C. Hsu, C.-H. Chang, H.-H. G. Tsai and D.-R. Hou, Eur. J. Org. Chem., 2010, 4771 CrossRef CAS PubMed; (i) R. Lahiri, H. P. Kokatla and Y. D. Vankar, Tetrahedron Lett., 2011, 52, 781 CrossRef CAS PubMed; (j) I. S. Kim, Q. R. Li, G. R. Dong, Y. C. Kim, Y. J. Hong, M. Lee, K.-W. Chi, J. S. Oh and Y. H. Jung, Eur. J. Org. Chem., 2010, 1569 CrossRef CAS PubMed; (k) M.-J. Chen and Y.-M. Tsai, Tetrahedron Lett., 2007, 48, 6271 CrossRef CAS PubMed; (l) S. R. Angle, D. Bensa and D. S. Belanger, J. Org. Chem., 2007, 72, 5592 CrossRef CAS PubMedand references cited therein. For a review on lentiginosine, see: (m) F. Cardona, A. Goti and A. Brandi, Eur. J. Org. Chem., 2007, 1551 CrossRef CAS PubMed; (n) F. M. Cordero, D. Giomi and A. Brandi, Curr. Top. Med. Chem., 2014, 14, 1294 CrossRef CAS.
- For recent syntheses of swainsonine, see: (a) P. Singh, S. K. Manna and G. Panda, Tetrahedron, 2014, 70, 1363 CrossRef CAS PubMed; (b) X.-G. Wang, A.-E. Wang and P.-Q. Huang, Chin. Chem. Lett., 2014, 25, 193 CrossRef CAS PubMed; (c) Q. R. Li, G. R. Dong, S. J. Park, Y. R. Hong, I. S. Kim and Y. H. Jung, Eur. J. Org. Chem., 2013, 4427 CrossRef CAS PubMed; (d) V. Dhand, J. A. Draper, J. Moore and R. Britton, Org. Lett., 2013, 15, 1914 CrossRef CAS PubMed; (e) G. Archibald, C. Lin, P. Boyd, D. Barker and V. J. Caprio, J. Org. Chem., 2012, 77, 7968 CrossRef CAS PubMed; (f) M.-J. Chen and Y.-M. Tsai, Tetrahedron, 2011, 67, 1564 CrossRef CAS PubMed; (g) H. G. Choi, J. H. Kwon, J. C. Kim, W. K. Lee, H. Eum and H.-J. Ha, Tetrahedron Lett., 2010, 51, 3284 CrossRef CAS PubMed; (h) D. J. Wardrop and E. G. Bowen, Org. Lett., 2011, 13, 2376 CrossRef CAS PubMed; (i) Y.-S. Tian, J.-E. Joo, B.-S. Kong, V.-T. Pham, K.-Y. Lee and W.-H. Ham, J. Org. Chem., 2009, 74, 3962 CrossRef CAS PubMed; (j) J. Louvel, F. Chemla, E. Demont, F. Ferreira and A. Perez-Luna, Org. Lett., 2011, 13, 6452 CrossRef CAS PubMed; (k) H. K. Zhang, S. Q. Xu, J. Zhuang, J. Ye and P. Q. Huang, Tetrahedron, 2012, 68, 6656 CrossRef CAS PubMed; (l) R. W. Bates and M. R. Dewey, Org. Lett., 2009, 11, 3706 CrossRef CAS PubMed; (m) S. J. Oxenford, S. P. Moore, G. Carbone, G. Barker, P. O'Brien, M. R. Shipton, J. Gilday and K. R. Campos, Tetrahedron: Asymmetry, 2010, 21, 1563 CrossRef CAS PubMed; (n) S. N. Murthy and Y. V. D. Nageswar, Synthesis, 2011, 755 CASFor a review of swainsonine syntheses, see: (o) L. Nin, H. Ba and A. Haji, Chin. J. Org. Chem., 2009, 29, 1354 Search PubMed; (p) S. G. Pyne, Curr. Org. Synth., 2005, 2, 39 CrossRef CAS; (q) A. E. Nemr, Tetrahedron, 2000, 56, 8579 CrossRef; (r) B. K. Lee, H. G. Choi, E. J. Roh, W. K. Lee and T. Sim, Tetrahedron Lett., 2013, 54, 553 CrossRef CAS PubMed.
- (a) S. P. Chavan, K. R. Harale, N. B. Dumare and U. R. Kalkote, Tetrahedron: Asymmetry, 2011, 22, 587 CrossRef CAS PubMed; (b) S. P. Chavan, N. B. Dumare, K. R. Harale and U. R. Kalkote, Tetrahedron Lett., 2011, 52, 404 CrossRef CAS PubMed; (c) S. P. Chavan, K. R. Harale and K. P. Pawar, Tetrahedron Lett., 2013, 54, 4851 CrossRef CAS PubMed; (d) S. P. Chavan and C. Praveen, Tetrahedron Lett., 2004, 45, 421 CrossRef CAS PubMed; (e) S. P. Chavan, N. B. Dumare and K. P. Pawar, RSC Adv., 2014, 4, 32594 RSC; (f) S. P. Chavan, L. B. Khairnar, P. C. Chavan and D. B. Kalbhor, Tetrahedron: Asymmetry, 2014, 25, 1246 CrossRef CAS PubMed; (g) S. P. Chavan, L. B. Khairnar, P. C. Chavan, N. B. Dumare and D. B. Kalbhor, Tetrahedron Lett., 2014, 55, 6423 CrossRef CAS PubMed.
- For reviews, see: (a) U. M. Lindström and P. Somfai, Synthesis, 1998, 109 CrossRef PubMed; (b) B. Zwanenburg and P. ten Holte, Top. Curr. Chem., 2001, 216, 93 CAS; (c) J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247 RSC; (d) X. E. Hu, Tetrahedron, 2004, 60, 2701 CrossRef CAS PubMed; (e) D. Tanner, Angew. Chem., Int. Ed. Engl., 1994, 33, 599 CrossRef PubMed; (f) H. M. I. Osborn and J. Sweeney, Tetrahedron: Asymmetry, 1997, 8, 1693 CrossRef CAS; (g) W. M. McCoull and F. A. Davis, Synthesis, 2000, 1347 CrossRef CAS PubMed; (h) I. D. G. Watson, L. Yu and A. K. Yudin, Acc. Chem. Res., 2006, 39, 194 CrossRef CAS PubMed; (i) A. Padwa and S. S. Murphree, ARKIVOC, 2006, 3, 6 Search PubMed; (j) D. S. Tsang, S. Yang, F. A. Alphonse and A. K. Yudin, Chem.–Eur. J., 2008, 14, 886 CrossRef CAS PubMed; (k) G. S. Singh, S. M. D'hooghe and N. De Kimpe, Chem. Rev., 2007, 107, 2080 CrossRef CAS PubMed; (l) C. Schneider, Angew. Chem., Int. Ed., 2009, 48, 2082 CrossRef CAS PubMed; (m) S. Stanković, S. D'hooghe, M. Catak, H. Eum, M. Waroquier, V. Van Speybroeck, N. De Kimpe and H.-J. Ha, Chem. Soc. Rev., 2012, 41, 643 RSC; (n) P. Lu, Tetrahedron, 2010, 66, 2549 CrossRef CAS PubMed; (o) T. Ishikawa, Heterocycles, 2012, 85, 2837 CrossRef CAS PubMed; (p) H. Ohno, Chem. Rev., 2014, 114, 7784 CrossRef CAS PubMed.
- (a) T. M. Shaikh and A. Sudalai, Tetrahedron: Asymmetry, 2009, 20, 2287 CrossRef CAS PubMed; (b) A. S. Kamal and R. Vangala, Tetrahedron, 2011, 67, 1341 CrossRef CAS PubMed; (c) M. K. Gurjar, L. Ghosh, M. Syamala and V. Jayasree, Tetrahedron Lett., 1994, 35, 8871 CrossRef CAS.
- (a) T. Oishi, T. Iwakuma, M. Hirama and S. Itô, Synlett, 1995, 404 CrossRef CAS PubMed; (b) S. Chooprayoon, C. Kuhakarn, P. Tuchinda, V. Reutrakul and M. Pohmakotr, Org. Biomol. Chem., 2011, 9, 531 RSC.
- D. B. Denny and S. T. Ross, J. Org. Chem., 1961, 27, 998 CrossRef.
- (a) H.-D. Ambrosi, W. Duczek, E. Gründemann, M. Ramm and K. Jähnisch, Liebigs Ann. Chem., 1994, 1013 CrossRef CAS PubMed; (b) K. S. Ajish Kumar, V. D. Chaudhari and D. D. Dhavale, Org. Biomol. Chem., 2008, 6, 703 RSC.
- K. W. C. Poon, K. M. Lovell, K. N. Dresner and A. Datta, J. Org. Chem., 2008, 73, 752 CrossRef CAS PubMed.
- (a) T. Bieg and W. Szeja, Synthesis, 1985, 76 CrossRef CASfor Pd catalyzed regioselective cleavage of vinyl aziridines please see: (b) Ref. 25b; (c) G. W. Spears, K. Nakanishi and Y. Ohfune, Synlett, 1991, 91 CrossRef CAS; (d) K. Fugami, K. Miura, Y. Morizawa, K. Oshima, K. Utimoto and H. Nozaki, Tetrahedron, 1989, 45, 3089 CrossRef CASfor hydrogenation of ester aziridines and C(2)–N, C(3)–N bond cleavage, please see: (e) Y. Lim and W. K. Lee, Tetrahedron Lett., 1995, 36, 8431 CrossRef CAS.
- C. Greck, F. Ferreira and J. P. Genêt, Tetrahedron Lett., 1996, 37, 2031 CrossRef CAS.
- G. Rassu, L. Auzzas, V. Zambrano, P. Burreddu, L. Pinna, L. Battistini, F. Zanardi and G. Casiraghi, J. Org. Chem., 2004, 69, 1625 CrossRef CAS PubMed.
- (a) P. J. Garegg and B. Samulesson, Synthesis, 1979, 469 CrossRef CAS; (b) H. B. Mereyala, P. M. Goud, R. R. Gadikota and K. R. Reddy, J. Carbohydr. Chem., 2000, 19, 1211 CrossRef CAS PubMed.
- (a) R. H. Grubbs, Handbook of Metathesis. In Applications in Organic Synthesis, WILEY-VCH, 2003, vol. 2 Search PubMed; (b) G. C. Fu and R. H. Grubbs, J. Am. Chem. Soc., 1992, 114, 7324 CrossRef CAS.
- S. C. Deshmukh, A. Roy and P. Talukdar, Org. Biomol. Chem., 2012, 10, 7536 CAS.
- H.-D. Ambrosi, W. Duczek, E. Gründemann, M. Ramm and K. Jähnisch, Liebigs Ann. Chem., 1994, 1013 CrossRef CAS PubMed.
- D. B. Denny and S. T. Ross, J. Org. Chem., 1961, 27, 998 CrossRef.
- K. Jähnisch, Liebigs Ann./Recl., 1997, 757 CrossRef PubMed.
- S.-B. Huang, J. S. Nelson and D. D. Weller, Synth. Commun., 1989, 19, 3485 CrossRef CAS PubMed.
- F. Sánchez-Sancho and B. Herradón, Tetrahedron: Asymmetry, 1998, 9, 1951 CrossRef.
- (a) D.-R. Hou, S.-Y. Hung and C.-C. Hu, Tetrahedron: Asymmetry, 2005, 16, 3858 CrossRef CAS PubMed; (b) L.-A. Watanabe, S. Haranaka, B. Jose, M. Yoshida, T. Kato, M. Moriguchi, K. Soda and N. Nishino, Tetrahedron: Asymmetry, 2005, 16, 903 CrossRef CAS PubMed.
- L. Battistini, F. Zanardi, G. Rassu, P. Spanu, G. Pelosi, G. G. Fava, M. B. Ferrari and G. Casiraghi, Tetrahedron: Asymmetry, 1997, 8, 2975 CrossRef CAS.
- W. H. Chiou, G. H. Lin and C. W. Liang, J. Org. Chem., 2010, 75, 174 CrossRef PubMed.
- (a) T. Oishi, T. Iwakuma, M. Hirama and S. Itô, Synlett, 1995, 404 CrossRef CAS PubMed; (b) S. Chooprayoon, C. Kuhakarn, P. Tuchinda, V. Reutrakul and M. Pohmakotr, Org. Biomol. Chem., 2011, 9, 531 RSC.
- S. C. Deshmukh, A. Roy and P. Talukdar, Org. Biomol. Chem., 2012, 10, 7536 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra06429e |
| This journal is © The Royal Society of Chemistry 2015 |