
Superior shape memory properties and microstructure evolution of poly(ether-b-amide12) elastomer enhanced by poly(ε-caprolactone)†
Abstract
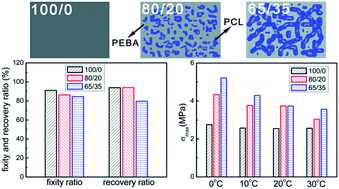
In this work, the superior shape memory properties and microstructure evolution of poly(ether-b-amide12) (PEBA) enhanced by poly(ε-caprolactone) (PCL) were investigated. The PEBA/PCL blends were prepared by the methods of solution mixing and compression molding. The representative phase-separated morphologies were observed by optical microscopy (OM). The recovery stress and shape memory properties of the samples were determined by dynamic mechanical analysis (DMA). It was found that high loadings of PCL (up to 35 wt%) only marginally worsened the recovery properties of the blends. However, the values of maximum recovery stresses increased greatly with PCL content in spite of deformation temperature, indicating an enhancement effect of PCL on the recovery stress of polymer blends. Moreover, in situ wide angle X-ray scattering (WAXS) investigation revealed the microstructure evolution during the shape memory process. It was notable that during the stretching process, the strain-induced crystallization of soft segments (PTMO) of PEBA shifted to lower strain due to the existence of the PCL phase. The present results provided a reference that the recovery stress could be greatly increased by adding another polymer to the shape memory polymer, which is vital to application in the biomedical field.
 Please wait while we load your content...
Please wait while we load your content...

