
Development of crystalline–amorphous parylene C structure in micro- and nano-range towards enhanced biocompatibility: the importance of oxygen plasma treatment time
M. Golda-Cepaa,
K. Engvallb and
A. Kotarba*a
aFaculty of Chemistry, Jagiellonian University, Ingardena 3, 30-060 Krakow, Poland. E-mail: kotarba@chemia.uj.edu.pl
bDepartment of Chemical Engineering and Technology, KTH Royal Institute of Technology, SE-100 44, Stockholm, Sweden
First published on 27th May 2015
Abstract
The crystalline–amorphous parylene C structure was fabricated by Chemical Vapour Deposited (CVD) and functionalised in the micro- and nano-range with the oxygen plasma treatment. The evolution of thermal stability, structure and surface biocompatibility of parylene C films as an effect of oxygen plasma treatment time were evaluated by means of thermogravimetric/differential thermal analysis (TG/DTA), X-Ray Diffraction (XRD) and cells adhesion tests (crystal violet assay, fluorescence microscopy). The results are epitomized by a crystalline–amorphous parylene C structural model. It was found that the time of oxygen plasma treatment is critical for adhesion of osteoblast cells with the optimum of 5–8 minutes.
Introduction
Polymers have been used for the last three decades in medical applications, such as coated transducers, cardiac assist devices and catheters. Recently, parylene C (poly(chloro-para-xylylene)) has drawn exceptional attention and is widely investigated in this context.1–3 Applied as a metal implants coating, parylene C provides a variety of benefits, such as low water permeability, durability and biocompatibility. Fabricated commercially semicrystalline parylene C layer has been proven as an excellent anticorrosive coating.4 The parylene C coatings on medical devices are stable in body environment and exhibit insignificant changes in their properties while adjusting to the physiological fluids. Electrochemical tests, show that an 8 μm layer makes an effective barrier for heavy metal ions release and dramatically inhibits associated processes of implant surface corrosion in the body.4 Continuous, dense, thin film of parylene C on metallic implants surface, is easily prepared via chemical vapour deposition (CVD). The film exhibits excellent mechanical properties required for metal implant applications, such as elasticity, strength, friction, durability and low permeability to water. The specific features of the materials are directly related to a hierarchical morphology typical for semicrystalline polymers. Particularly, the CVD parylene C structure can be considered as a crystalline–amorphous composite with nano and micro crystalline blocks.5 Whereas, the mechanical strength and low water permeability are associated with the dense crystalline structure, the amorphous part can easily absorb mechanical and thermal shocks. Additionally, parylene C is one of the most bio-accepted coatings for permanently implanted devices, e.g. stents, defibrillators, pacemakers, as well as guide wires, sensors and probes. It is important to emphasise that the clinical success of orthopaedic implants coating relies on the quick and efficient formation of bone tissue at an implanted surface. Therefore, it is extremely important to ensure specific surface properties,6 required for biointegration, which can be discussed in terms of osteoblast attachment, responsible for the bone tissue formation. It was previously observed that this complex process is governed by the surface free energy of the material7 and its topography.8 The early interactions of body fluids with a surface of artificial material play a fundamental role in the cell adhesion process, the differentiation and the eventual tissue formation on the interface. Physicochemical cell–surface interactions, resulting from inherent chemistry and topography, govern and drive osteoblast adhesion and maturation processes. Cells do not adhere uniformly to the surface, but at local surface sites, called focal adhesions, which are generated by protein clusters, mediated by integrins.17 It was previously reported, that focal adhesion sites, preferably, are created on a surface rich in oxygen containing groups9 and on surface with nano-topography.10 These influence cellular behaviour by alteration of the number of available integrin binding sites.One of the currently used techniques for surface engineering is oxygen plasma, created by an electrical glow discharge in vacuum, forming reactive oxygen species, interacting with the polymeric material surface. Several papers11–13 indicate that plasma surface etching can be well adapted for the preparation of biomaterials surfaces, due to its reproducibility, adjustability, as well as its ability to provide the required cleanness of the treated material.14 The plasma technique offers the possibility of tuning the chemical composition and properties of a surface homogeneously, and thus; improve the overall performance of the material. Moreover, it can be applied to any type of polymeric material and surface geometry. Due to its many advantages, it appears, nowadays, as one of the most versatile process in biomaterials engineering upon plasma treatment, oxygen-containing groups are formed on the surface (–C(O)H, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –COOH), providing the adhesion centres for the osteoblast cells. Additionally, the modification can be precisely controlled by adjusting the oxygen partial pressure, the power and the time of exposure.15 Since polymeric materials interact with plasma differently, depending on its chemical composition and crystallinity degree, as well as surface structure, the treatment parameters have to be carefully adjusted for each particular polymer.
O, –COOH), providing the adhesion centres for the osteoblast cells. Additionally, the modification can be precisely controlled by adjusting the oxygen partial pressure, the power and the time of exposure.15 Since polymeric materials interact with plasma differently, depending on its chemical composition and crystallinity degree, as well as surface structure, the treatment parameters have to be carefully adjusted for each particular polymer.
In our previous papers, the surface of parylene C was functionalized by formation of surface nanoroughness and specific chemical groups via oxygen plasma treatment,15,16 an as a result, enhanced the osteoblast adhesion.17 It has to be emphasised that the parameters of the mild oxygen plasma modification were controlled in a precise way. Too long exposure leads to excessive introduction of oxygen into the polymer, which may result in weakening of mechanical strength and reducing thermal stability, crucial for the surgical procedure and thermal sterilization, respectively.18 The parylene C coating is stable over a wide temperature range (−200 °C to 200 °C), allowing the coated items to be autoclaved. It should be mentioned, however, that there are several reports discussing autoclaving as a suitable method for parylene C sterilization.6,19 Another issue concerns, if the plasma modification and insertion of oxygen into the polymer chains influence the parylene C structure, the stability and biocompatibility. Whatever sterilization process (ethylene oxide, thermal sterilization, irradiation), it must be integrated into any concept of novel biomaterials fabrication and their surface engineering.
As described above, while treated with oxygen plasma, several key factors are essential for practical applications of parylene C. The factors can be analysed in terms of structure and surface changes, as well as biofunctionalization, forming a set of interlaced issues. These interlaced issues have, to our best knowledge, so far, never been combined into an integral study of this material. Also, to be emphasized, since oxygen plasma is becoming widely used for polymer surface functionalization, the results from such a study are of a more general notion. Most of polymers used, as biomaterials are semi-crystalline and, due to various chemical structures, the susceptibility of amorphous and crystalline domains to oxygen plasma exposure differs substantially.
The aim of the presented study was to investigate the evolution of thermal stability, structure and biocompatibility of parylene C films as an effect of oxygen plasma treatment time. Each of these points was investigated with the dedicated method: thermogravimetric/differential thermal analysis (TG/DTA), X-Ray Diffraction (XRD) and cells adhesion tests (crystal violet assay, fluorescence microscopy).
Materials and methods
Sample preparation
Parylene C (8 μm of thickness) films were prepared by Chemical Vapour Deposition (CVD) technique, provided by ParaTech Coating Scandinavia AB. The dimer of chloro-para-xylylene was used as a precursor and heated up to 690 °C for decomposition to monomeric form. The monomers in vapour phase were spontaneously deposited and polymerized at room temperature at 10−3 mbar.20 The thickness of the coating was controlled by the deposition time. The samples were cleaned in isopropyl alcohol and distilled water before further investigations.To evaluate the influence of parylene C functionalization on its thermal stability, three modification procedures were applied: oxygen plasma treatment, autoclaving and heating at ambient conditions.
In order to modify the parylene C surface, oxygen plasma treatment was carried out using a Diener electronic Femto plasma system (Diener Electronic GmbH, Nagold, Germany) at 50 W and an oxygen partial pressure of 0.2 mbar. The varied parameter was the time of exposure to the plasma, which was in the range of 1–60 min. During the plasma treatment, the surface temperature of parylene C increases to 70–80 °C, close to the glass transition temperature.
The most popular quick disinfection method in surgery is standard steam autoclave sterilization.18 Therefore, as a reference, samples of autoclaved parylene C films were prepared. The procedure was performed in a Nüve OT 40L laboratory steam sterilizer with the following parameters: 121 °C, 20 min and 1 atm.
Thermogravimetric analysis (TGA/DTA)
The properties of parylene C samples were analysed using a TGA/DTA Mettler-Toledo apparatus. The samples of about 10 mg were placed in an open alumina crucibles. The experiments were carried out in a temperature range of 30–600 °C with a heating rate of 6.5 °C min−1 at an Ar flow of 50 cm3 min−1.X-ray diffraction (XRD)
The XRD patterns of parylene C foils were recorded by a Rigaku MiniFlex diffractometer with Cu Kα radiation at 10 mA and 10 kV, 2θ step scans of 0.02 and a counting time of 1 s per step. The diffraction measurements were performed in the characteristic region of 2θ = 3–40°, the crystallite size was estimated by the Sherrer's formula and the crystallinity was calculated from the areas under the amorphous and crystalline peaks, as described by Hyun.18Cell culture
Parylene C disks, used in the cell culture study, were sterilized by exposure to UV radiation. For evaluation of cell adhesion, the MG-63 human osteosarcoma cell line was employed. Both unmodified and treated with oxygen plasma were used in the experiments. Cells were grown in Dulbecco's Modified Eagle's Medium (DMEM), supplemented with 2 mM glutamine and 10% fetal bovine serum (FBS), for 24 h, 48 h and 72 h with a control sample (cells without parylene C film) in 24-well tissue culture plates.Cells fluorescent staining
The changes in cells morphology and adhesion rate on parylene C surfaces were investigated using an Olympus IX51 fluorescence microscope, equipped with a XC10 camera. Cells were fixed in 4% PFA, permeabilized in 0.5% Triton-X100. Cells cytoskeleton was stained with Alexa Fluor 488 dye (Molecular Probes) and nuclei with DAPI (Molecular Probes). After observation, the colour images of cytoskeleton and nuclei were combined.Cells adhesion test
The different affinity of the cells to the investigated parylene C surfaces was determined by their adhesion rate. In order to quantify the effect of plasma treatment, standard crystal violet assay procedure was applied. Cells were seeded in 24-well plates at a density of 4 × 103 per well. The tests were performed for 24, 48 and 72 h, and wells without parylene C film were used as a control. After the time intervals, adherent cells were washed in phosphate buffer saline (PBS), fixed in 4% paraformaldehyde (PFA), and stained with crystal violet. After extensive washings with distilled water, plates were allowed to dry. The amount of crystal violet absorbed by cells was dissolved in methanol (99.5%). The absorbance was measured, using a microplate reader (Infinite 200 M PRO NanoQuant, Tecan, Switzerland) at λ = 570 nm.At least three independent cells adhesion tests were completed for each polymer surface, resulting in a standard deviation for OD570 of 0.1.
Results
Oxygen plasma treatment affects several properties related to the surface of parylene C, such as thermal stability, structure and biocompatibility. Since these parameters influence each other, it is very important with a parallel evaluation to gain the overall picture. At first, the results are presented separately and then discussed and integrated into a comprehensive structural model, combining the changes in micro and nano domains of crystalline–amorphous parylene C composite.TGA/DTA
Representative thermogravimetrical results of unmodified and oxygen plasma modified parylene C are compared in Fig. 1–3. Fig. 1 shows the typical parylene C TGA curve, including also three characteristic ranges of temperature, representing different material transitions, specifically: 60–90 °C – glass transition (Tglass), 293 °C – melting (Tmelt)19 and 473.2 °C – degradation (Tdegrad). As one of the aims of this study is to evaluate the thermal stability of oxygen plasma treated parylene C in a biomedical context, the range of steam sterilisation temperature (in orange) at around 130 °C is also marked in Fig. 1. This temperature range is well below the parylene C Tmelt, directly indicating that steam sterilization can be safely applied for this material.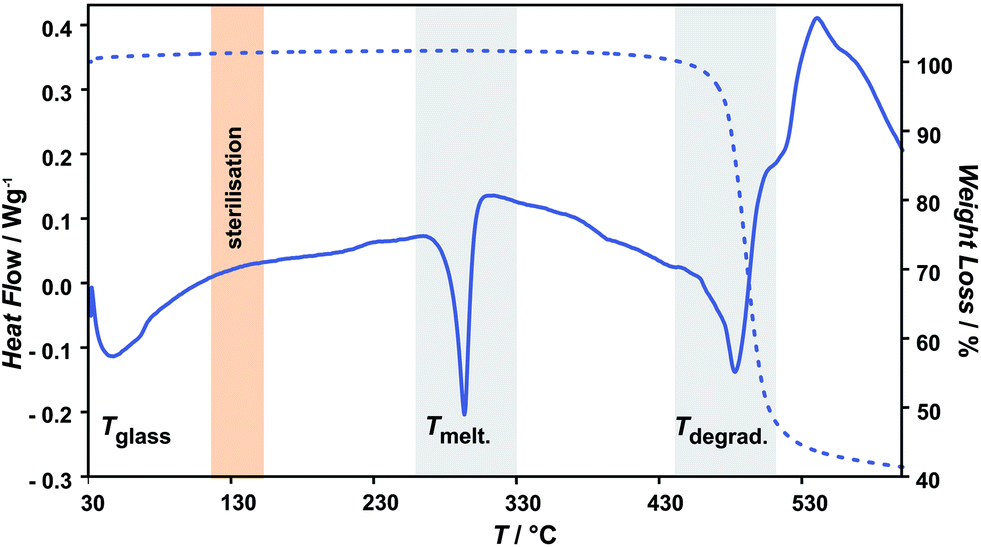 | ||
| Fig. 1 Representative thermogravimetrical curves of unmodified parylene C with characteristic temperature ranges: melting (Tmelt) and degradation (Tdegrad) (in grey) and steam sterilization (in orange). | ||
The TG curves in Fig. 2 present the Tdegrad for unmodified and oxygen plasma treated parylene C (3–60 min). The degradation temperature are between 465 and 471 °C for the examined samples, revealing a high thermal stability of the oxygen plasma treated samples in agreement with values reported in the literature.23 The observed rapid decrease of molecular weight, for all of the polymer films, in this temperature range, can be attributed to continuous breaking of C–C bonds, when longer-chain polymeric molecules are transformed into volatile shorter molecules.19
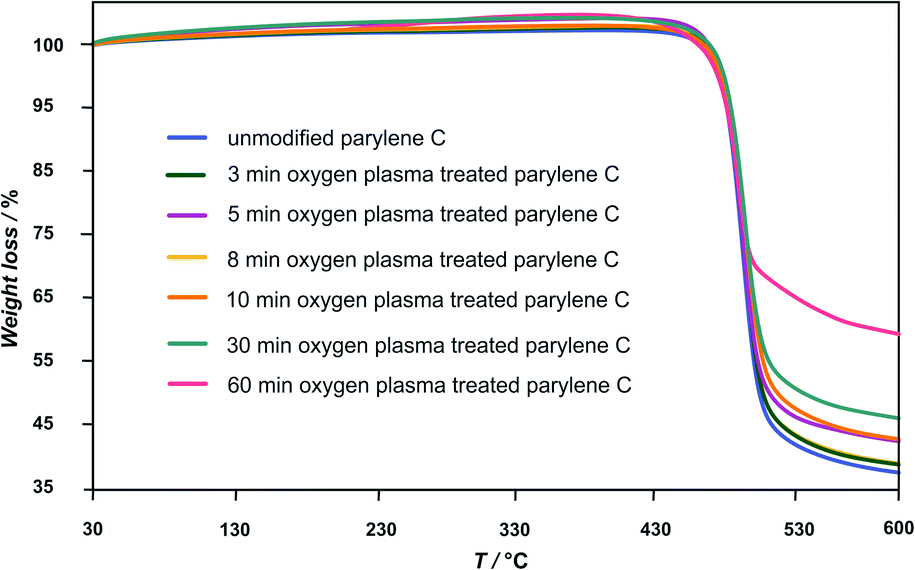 | ||
| Fig. 2 The TG curves presenting the Tdegrad for unmodified and oxygen plasma treated parylene C (3–60 min). | ||
As a consequence, the total degradation of the polymer structure takes place in this temperature range. The longer the oxygen plasma treatment time, the lower the observed weight loss of the polymer sample: 62 ± 2% for unmodified and 3 min, 5 min, 8 min, oxygen plasma treated samples, 57% for 10 min, 54% for 30 min and 41% for 60 min.
This finding indicates the complex structural changes in crystalline–amorphous parylene C composite revealed by XRD (see below).
Changes in the melting temperature (Tmelt) are presented in Fig. 3. A characteristic trend in Tmelt, as a function of plasma exposure time, is observed for the unmodified, 8 min and 60 min oxygen plasma modified samples (Fig. 3A). A more detailed examination including all samples, is shown in Fig. 3B, illustrated by the plotted results for Tmelt vs. plasma exposure time. The Tmelt of unmodified parylene C is 298.7 °C and upon exposure to plasma the Tmelt of modified parylene C films increase gradually from 300 °C for 3 min plasma treatment, peaking at 301.4 °C for 8 min. Longer exposure to plasma causes gradual decrease of Tmelt, to 293.7 °C for 10 min, 291.3 °C for 30 min and finally to 288.2 °C for 60 min exposure. The temperature shift (ΔTmelt ≈ 10 °C) indicates clearly significant bulk changes in the polymer structure upon longer plasma treatment. For comparison, TGA/DTA tests with a reference sample of autoclaved parylene C foils were performed. The observed Tmelt for autoclaved parylene C is 298.0 °C, thus essentially the same, within the experimental limits, as for the unmodified parylene C foils. From the observations, it can be inferred that oxygen plasma treatment more profoundly affects the stability than thermal sterilization. A further insight into the structural changes can be gained from XRD measurements, described in the next section.
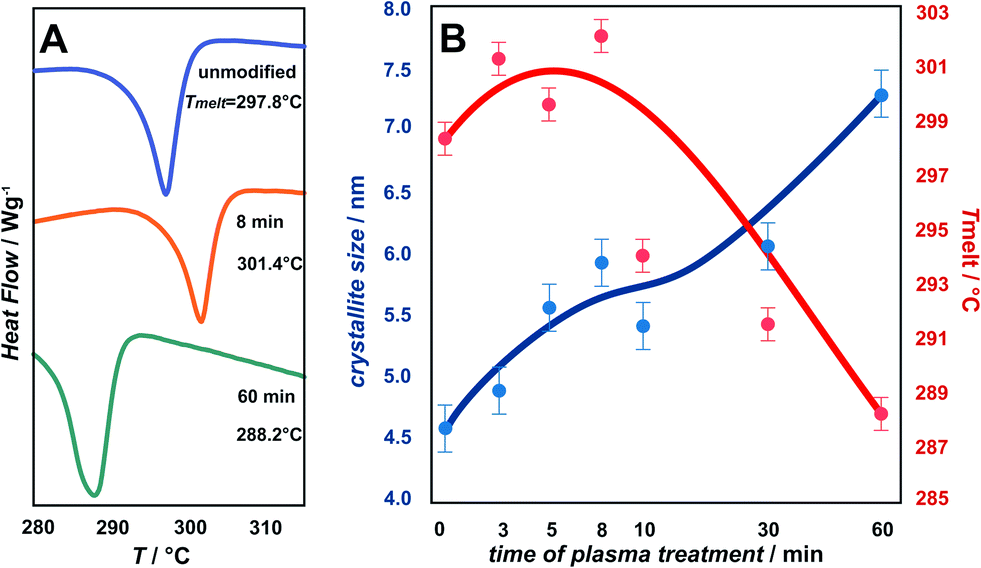 | ||
| Fig. 3 Changes in the melting temperature (Tmelt). For the unmodified, 8 min and 60 min oxygen plasma modified samples (A) together with the detailed plot of the Tmelt (in red) and crystallites size (in blue) for all of the examined samples (B). | ||
XRD
The degree of polymer crystallinity plays an important role in the process of the oxygen plasma treatment. The chain mobility of the crystalline part of parylene C is restricted due to its densely packed structure, limiting the penetration of plasma-generated reactive oxygen species into the material bulk. The changes in crystalline–amorphous components of the parylene C were examined by XRD.Fig. 4 displays the dependency of oxygen plasma exposure time on the evolution of XRD peaks for parylene C film. The diffraction patterns show a distinct maxima at 2θ ≈ 13.7°, which can be indexed within a space group P1.21 The second broad maxima, observed for higher 2θ values in the range 20–40°, can be attributed to amorphous parts of the crystalline–amorphous parylene C composite. The maximum, assigned to the (020) crystallographic plane, increases upon oxygen plasma exposure. This can be explained in terms of an increase in crystallite size, as presented in Fig. 3B (in blue) together with the changes in Tmelt. The crystallite size, calculated using the Scherrer formula, for the unmodified parylene C is 4.6 nm. This size gradually increases to 4.9 nm and subsequently to 7.2 nm for the 3 min and for the 60 min plasma treated samples, respectively. The largest crystallites were found for the autoclaved parylene C. Nevertheless, also in this case, the crystallite size of about 9.5 nm is still very small. The changes in size of crystallite and amorphous regions can be quantified in terms of the degree of polymer crystallinity. For unmodified parylene C, the observed crystallinity is 27%, as also obtained for the samples treated for 3 and 5 minutes. After 8 minutes of treatment, the crystallinity decreases to 23%. Thereafter, a crystallinity of 29% is obtained for the samples modified for 10 min. For the modification time of 30 and 60 min, the crystallinity degree dramatically decreases to 17% and thereafter again increases to 23%, respectively. Autoclaved parylene C films exhibit the lowest crystallinity of all of the examined samples of 8%. The observed results indicate that, at first, the balance between the ratio of crystalline![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :amorphous parts remain the same as for the unmodified samples. However, after exposure to plasma for 8 min a secondary amorphisation occurs. This happens when particle shower, oxidation and vacuum ultraviolet radiation (VUV) from a plasma cause crosslinking and degradation of polymer surface-near layers.25 The crystallinity does not change in a monotonous way with the oxygen plasma treatment time, thus there is no clear correlation between crystallinity and the biocompatibility of parylene C. The reason is the competition between three parallel–consecutive processes: etching of amorphous and crystalline regions and secondary amorphisation with significantly different kinetics. The increase in the degree of crystallinity upon oxygen plasma treatment, observed for the samples treated for 10 and 60 min is due to preferential etching of the amorphous phase, whereas crystalline regions are more etch-resistant. The explanation for the etch selectivity in semi-crystalline polymers, is most likely the lower permeability of plasma reactive species through the dense, crystalline regions. The diffusion of reactive particles into a polymer matrix is known to be one of the most important factors in the functionalization via chemical etching processes.25
:amorphous parts remain the same as for the unmodified samples. However, after exposure to plasma for 8 min a secondary amorphisation occurs. This happens when particle shower, oxidation and vacuum ultraviolet radiation (VUV) from a plasma cause crosslinking and degradation of polymer surface-near layers.25 The crystallinity does not change in a monotonous way with the oxygen plasma treatment time, thus there is no clear correlation between crystallinity and the biocompatibility of parylene C. The reason is the competition between three parallel–consecutive processes: etching of amorphous and crystalline regions and secondary amorphisation with significantly different kinetics. The increase in the degree of crystallinity upon oxygen plasma treatment, observed for the samples treated for 10 and 60 min is due to preferential etching of the amorphous phase, whereas crystalline regions are more etch-resistant. The explanation for the etch selectivity in semi-crystalline polymers, is most likely the lower permeability of plasma reactive species through the dense, crystalline regions. The diffusion of reactive particles into a polymer matrix is known to be one of the most important factors in the functionalization via chemical etching processes.25
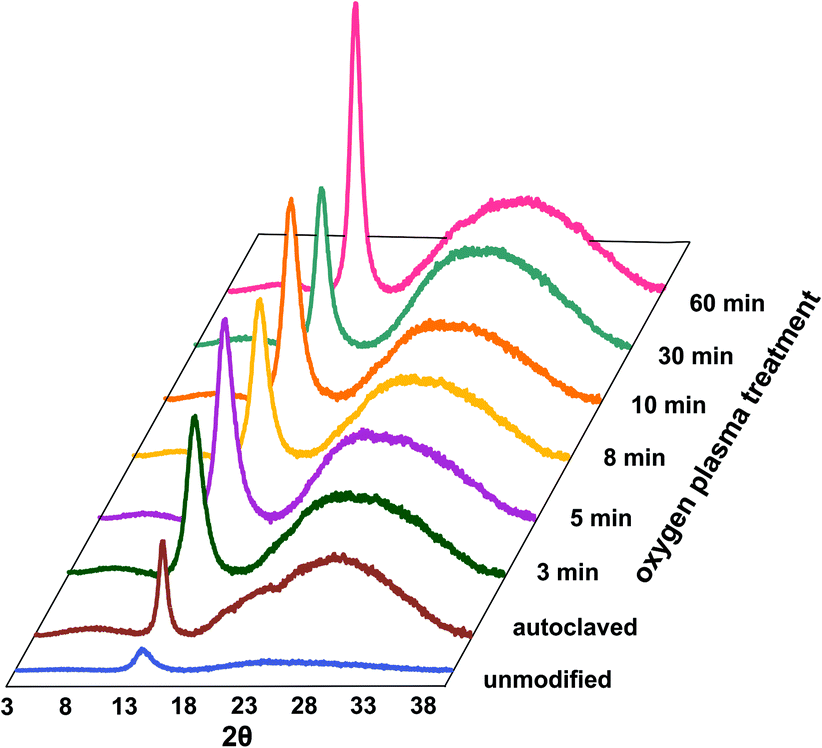 | ||
| Fig. 4 Dependence of oxygen plasma exposure time on evolution of XRD peaks for parylene C. | ||
Summarizing the results from the TG/DTA and XRD an overall model of oxygen plasma induced changes for parylene C can be proposed, as presented in Fig. 5. Apart from surface chemical modification, forming functional oxygen groups, during the first minutes of oxygen plasma treatment, as demonstrated in our previous study,16 also surface etching of amorphous phase takes place, leading to the formation of cavities in the range of 200 nm. Simultaneously, the empty space, as a result of the etching, allows the polymeric chains to reorganize, resulting in an increase in crystallite size as observed by XRD.
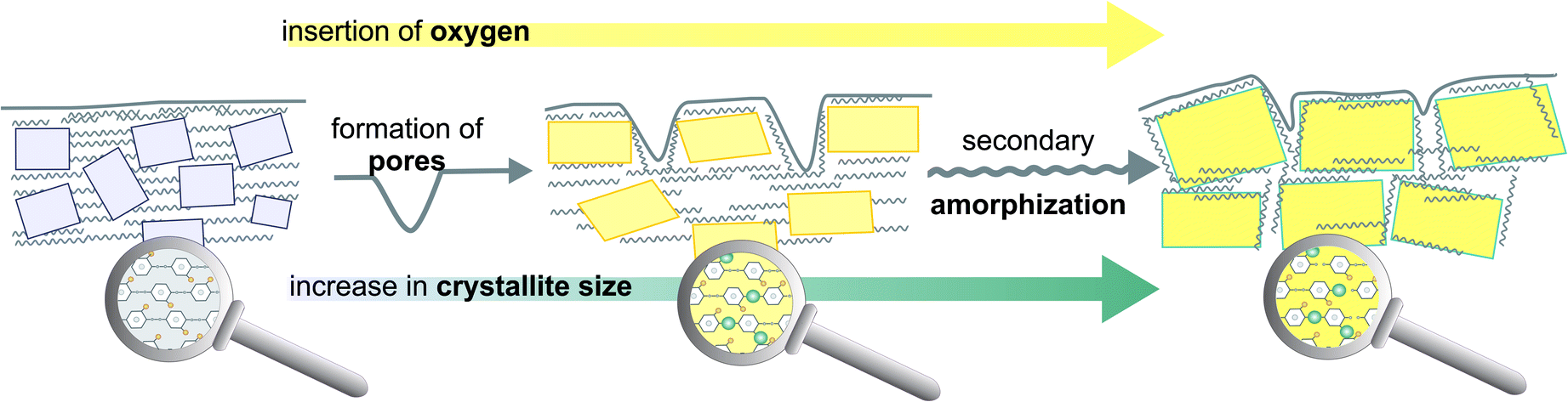 | ||
| Fig. 5 Structural model combining micro and nano domains of crystalline–amorphous parylene C composite illustrating the main features of oxygen plasma induced changes. | ||
MG-63 microscopic observations and adhesion test
Cell adhesion to synthetic surfaces is crucial for many biomedical and biotechnological applications. In addition to anchoring cells, adhesive interactions activate various intracellular signalling pathways that direct cell viability, proliferation, and differentiation.Keselowsky et al.22 demonstrated that surface chemistry modulates focal adhesion composition and signalling. Osteoblasts preferentially bind to –COOH and –OH surface groups. These findings offer a practical guideline for the engineering of surfaces that elicit specific cellular responses for potential mechanism for the diverse cellular responses to biomaterial surfaces. Fig. 6 presents fluorescent microscopy observations after 72 h of cells incubation. The results clearly show superior cell attachment to plasma-functionalized parylene C, compared to what is observed for the unmodified surface. For the modified surface, the cells were evenly spread, displaying a closely packed congruent structure. In case of the reference, with regard to the unmodified parylene C film, the cells were separated from each other and the surface coverage was uneven. The adhesion of the cells to the parylene C films, monitored for 3 days, is presented as quantified adhesion rates in Fig. 7. For all of the examined samples, a reproducible trend is observed. The poorest adhesion is determined for the unmodified parylene C, while after a short time of plasma modification, the number of adhered cells, starts to increase. The optimal time of plasma treatment is in the range of 5–8 min, as for these modification times, cells adhered in the largest amount after the incubation time of 24 and 48 h.
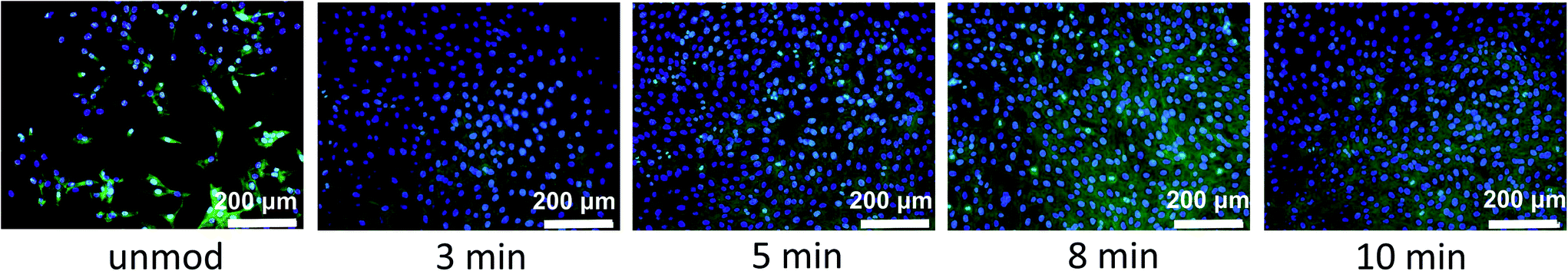 | ||
| Fig. 6 Representative fluorescent microscopy images of MG-63 cells after 72 h incubation on parylene C surfaces treated with oxygen plasma for different time. | ||
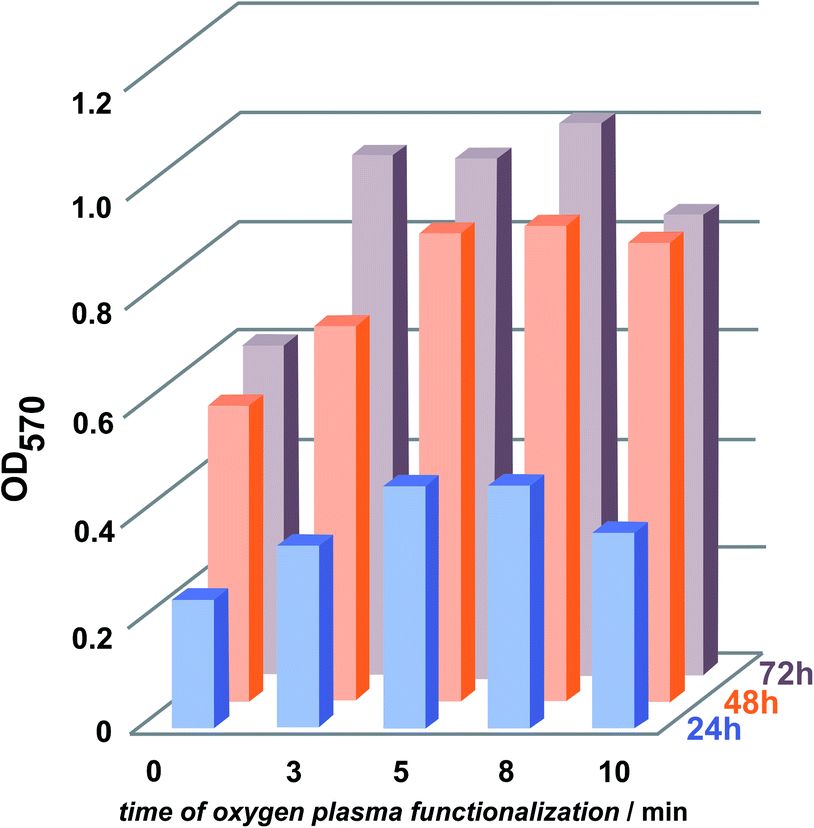 | ||
| Fig. 7 Quantification of adhesion rate of MG-63 cells after 24, 48 and 72 h to the oxygen plasma modified parylene C surfaces. | ||
After 72 h, there is a slight increase in the attached cells for the 8 min modified surfaces. The trend is quite similar to the changes observed with XRD for the crystallites growth-with up to 8 min of plasma modification, the size of the crystals increase and after 10 min it decreases. It has to be emphasised however, that the optimal time of the plasma treatment have to be adjusted for the specific parylene C material, e.g. the ratio of crystalline/amorphous regions, film thickness and the substrate material can vary24,26 and thus, affect the optimal modification parameters in each case. Studies on osteoblasts suggest that the attachment of the cells to crystalline surfaces supposed to be lower than for amorphous surfaces.27,28
However, it has to be emphasised that the results for cell adhesion are not only caused by crystallinity of the films but is also affected by the chemistry and topography of the surface. The observed adhesion rates are therefore a consequence of all 3 factors.
Conclusions
A series of parylene C films, prepared by the CVD method, was functionalized via oxygen plasma treatment with various exposure times. The evolution of thermal stability, structure and biocompatibility of the prepared films, as an effect of oxygen plasma treatment time, was investigated by TG/DTA, XRD and osteoblast cells adhesion tests, respectively. It is concluded that the beneficial effect of oxygen plasma treatment on parylene C properties strongly depends on the time of exposure to oxygen reactive species.For the first time, a comprehensive structural model of the parylene C surface evolution in the micro- and nano-range upon interaction with oxygen plasma is proposed. At first stage, the amorphous regions of crystalline–amorphous composite are etched, resulting in the increase in Tmelt and crystallite size. Overexposure to oxygen plasma, significantly, alters the bulk properties of parylene C, simultaneously as the increase in the crystallite size by the secondary amorphization is observed. These changes are reflected in the degree of parylene C crystallinity. The preferred time of oxygen plasma treatment of parylene C, stimulating adhesion of osteoblast cells, is 5 to 8 minutes.
The practical implications of the obtained results, underline the importance of the oxygen plasma treatment time as a critical parameter for optimising enhanced biocompatibility of a parylene C coating. Since the assumptions for the model are of a general nature, the obtained results can be easily extended for other semi-crystalline polymers of biomedical importance.
Acknowledgements
This research project was operated within the Foundation for Polish Science Ventures Programme, co-financed by the EU European Regional Development Fund. Decision number: Ventures: 2012-10/2.References
- E. Delivopoulos, M. M. Ouberai, P. D. Coffey, M. J. Swann, K. M. Shakesheff and M. E. Welland, Colloids Surf., B, 2015, 126, 169 CrossRef CAS PubMed.
- E. Delivopoulos, A. F. Murray, N. K. MacLeod and J. C. Curtis, Biomaterials, 2009, 30, 2048 CrossRef CAS PubMed.
- C. P. Unsworth, H. Holloway, E. Delivopoulos, A. F. Murray, M. C. Simpson, M. E. Dickinson and E. S. Graham, Biomaterials, 2011, 32, 6541 CrossRef CAS PubMed.
- M. Cieślik, M. Kot, W. Reczyński, K. Engvall, W. Rakowski and A. Kotarba, Mater. Sci. Eng., C, 2012, 32, 31 CrossRef PubMed.
- S. Isoda, T. Ichida, A. Kawaguchi and K. I. Katayama, Bull. Inst. Chem. Res., Kyoto Univ., 1983, 61, 222 CAS.
- O. Grinberg, M. Natan, A. Lipovsky, A. Varvak, H. Keppner, A. Gedanken and E. Banin, J. Mater. Chem. B, 2015, 3, 59 RSC.
- S. B. Kennedy, N. R. Washburn, C. G. Simon and E. J. Amis, Biomaterials, 2006, 27, 3817 CrossRef CAS PubMed.
- G. Zhao, A. L. Raines, M. Wieland, Z. Schwartz and B. D. Boyan, Biomaterials, 2007, 28, 2821 CrossRef CAS PubMed.
- Y. Arima and H. Iwata, Biomaterials, 2007, 28, 3074 CrossRef CAS PubMed.
- R. V. Goreham, A. Mierczynska, L. E. Smith, R. Sedev and K. Vasilev, RSC Adv., 2013, 3, 10309 RSC.
- F. Poncin-Epaillard, T. Vrlinic, D. Debarnot, M. Mozetic, A. Coudreuse, G. Legeay, B. El Moualij and W. Zorzi, J. Funct. Biomater., 2012, 3, 528 CrossRef CAS PubMed.
- T. Jacobs, R. Morent, N. De Geyter, P. Dubruel and C. Leys, Plasma Chem. Plasma Process., 2012, 32, 1039 CrossRef CAS.
- A. Nandakumar, Z. T. Birgani, D. Santos, A. Mentink, N. Auffermann, K. van der Werf, M. Bennink, L. Moroni, C. van Blitterswijk and P. Habibovic, Biofabrication, 2013, 5, 015006 CrossRef CAS PubMed.
- S. Cheruthazhekatt, M. Černák, P. Slavíček and J. Havel, J. Appl. Biomed., 2010, 8, 55 CrossRef CAS PubMed.
- M. Gołda, M. Brzychczy-Włoch, M. Faryna, K. Engvall and A. Kotarba, Mater. Sci. Eng., C, 2013, 33, 4221 CrossRef PubMed.
- M. Gołda-Cępa, N. Aminlashgari, M. Hakkarainen, K. Engvall and A. Kotarba, RSC Adv., 2014, 4, 26240 RSC.
- R. A. Weinstein, D. R. Linkin, C. Sausman, L. Santos, C. Lyons, C. Fox, L. Aumiller, J. Esterhai, B. Pittman and E. Lautenbach, Clin. Infect. Dis., 2005, 41, 1014 CrossRef PubMed.
- Q. He, J. Q. Liu, B. Yang, X. Chen and C. S. Yang, Surf. Coat. Technol., 2014, 252, 120 CrossRef CAS PubMed.
- C. P. Tan and H. G. Craighead, Materials, 2010, 3, 1803 CrossRef CAS PubMed.
- J. Hyun, Polymer, 2001, 42, 6473 CrossRef CAS.
- B. J. Kim, B. Chen, M. Gupta and E. Meng, J. Micromech. Microeng., 2014, 24, 065003 CrossRef.
- B. G. Keselowsky, D. M. Collard and A. J. García, Surface chemistry modulates focal adhesion composition and signaling through changes in integrin binding, Biomaterials, 2004, 25(28), 5947–5954 CrossRef CAS PubMed.
- A. Kahouli, A. Sylvestre, F. Jomni, B. Yangui and J. Legrand, Appl. Phys. A, 2012, 106, 909 CrossRef CAS.
- K. Fukuda, T. Suzuki, D. Kumaki and S. Tokito, Phys. Status Solidi A, 2012, 209, 2073 CrossRef CAS PubMed.
- J. Friedrich, The Plasma Chemistry of Polymer Surfaces, Wiley-VCH, 2012 Search PubMed.
- C. Chindam, N. M. Wonderling, A. Lakhtakia, O. O. Awadelkarim and W. Orfali, Appl. Surf. Sci., 2015, 345, 145 CrossRef CAS PubMed.
- N. R. Washburn, K. M. Yamada, C. G. Simon, S. B. Kennedy and E. J. Amis, Biomaterials, 2004, 25, 1215 CrossRef CAS PubMed.
- G. Balasundaram, M. Sato and T. J. Webster, Biomaterials, 2006, 27, 2798 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |