
Diopside modified porous polyglycolide scaffolds with improved properties
Pei Feng†
b,
Xiaoning Guo†cd,
Chengde Gaob,
Dan Gaoaf,
Tao Xiaocd,
Xiong Shuaie,
Cijun Shuai*bd and
Shuping Peng*af
aHunan Provincial Tumor Hospital and the Affiliated Tumor Hospital of Xiangya School of Medicine, Central South University, Changsha, China 410013. E-mail: shuping@csu.edu.cn; Fax: +86-731-88879044; Tel: +86-731-84805412
bState Key Laboratory of High Performance Complex Manufacturing, Central South University, Changsha, China 410083. E-mail: shuai@csu.edu.cn; Fax: +86-731-88879044; Tel: +86-731-88879351
cDepartment of Orthopedics, The Second Xiangya Hospital, Central South University, Changsha, China 410011
dOrthopedic Biomedical Materials Institute, Central South University, Changsha, China 410083
eState Key Laboratory of Powder Metallurgy, Central South University, Changsha, China 410083
fSchool of Basic Medical Science, Central South University, Changsha, China 410078
First published on 12th June 2015
Abstract
Polyglycolide (PGA) is considered an attractive candidate for bone regeneration because of its good biodegradability and biocompatibility. However, its insufficient mechanical strength and inadequate bioactivity limit its applications. In this study, diopside (DIOP) was incorporated into PGA scaffolds to enhance the mechanical and biological properties. The porous scaffolds were fabricated via selective laser sintering (SLS). The effect of the DIOP content on the microstructure, mechanical properties, bioactivity as well as cytocompatibility of the porous scaffolds was studied. The results showed that DIOP particles were homogenously distributed within the PGA matrix, which contained up to 10 wt%. This led to an improvement of 171.2% in compressive strength and 46.2% in compressive modulus. In vitro studies demonstrated that the highest apatite forming ability was obtained on the scaffold surfaces with the highest amount of DIOP after soaking in simulated body fluid (SBF), suggesting that the bioactivity of the scaffolds increased with increasing DIOP. In addition, a cytocompatibility study showed that the scaffolds exhibited a higher degree of cell attachment, growth and differentiation than the pure PGA scaffolds. These indicated that the PGA scaffolds modified with DIOP possessed suitable properties which could be used for bone tissue regeneration.
1 Introduction
PGA has attracted increasing attention for its use as a bone scaffold material in the last 20 years due to the combined advantages of good biocompatibility and controllable degradation rates.1,2 It can be easily processed into a variety of porous structures via 3D printing, porogen leaching, electrospinning method, and so on.3–5 Moreover, the hydrophilic character of PGA can have benefits for cell adhesion, migration, proliferation and differentiation.4–6 However, scaffolds fabricated from PGA suffer from relatively low mechanical properties and limited bioactivity, which are disadvantages for bone regeneration.7 Furthermore, PGA degradation in biological environment can cause pH decrease due to the release of acidic degradation products (glycolic acid), which may lead to inflammatory response.8Considerable efforts are focused on improving the mechanical and biological properties of the biopolymer through the incorporation of bioactive inorganic phases such as tricalcium phosphate (TCP),2,9 hydroxyapatite (HAP)10–12 as well as bioactive glasses (BG).13–15 Cao et al.2 fabricated the PGA/β-TCP porous composite scaffolds via a solvent casting/particulate leaching method and confirmed the strong ability for mineralization and osteogenesis of the scaffolds. Dong et al.10 prepared the porous nano-HAP/polyurethane (PU) composite scaffold by a foaming method and found that nano-HAP could improve the bioactivity of scaffold in terms of cytocompatibility and biocompatibility. Lei et al.15 produced the poly(ε-caprolactone) (PCL)-BG composites and found that the addition of BG led to an increase in elastic modulus and apatite crystals.
DIOP (CaMgSi2O6) is a Ca-, Mg- and Si-containing bioceramic, which has generated great interest because of the favorable osteostimulation, excellent biocompatibility as well as apatite mineralization ability.16–18 Its mechanical properties such as toughness and strength are superior to those of calcium phosphate ceramics, glasses and glass ceramics.19,20 Previous studies demonstrated that DIOP had excellent apatite formation and could induce bone formation.21,22 Furthermore, it can release Ca2+, Si4+ and Mg2+ and produce an elevated local pH, which neutralizes the acidic products of PGA and stimulates osteoblasts to grow and proliferate.23,24 Based on these considerations, it can be predicted that the introduction of DIOP into PGA may enhance the mechanical properties and bioactivity.
This study investigated the incorporation of different amount of DIOP into the PAG scaffolds to enhance the mechanical and biological properties. The porous scaffolds were prepared via a home-made SLS machine. Scanning electron microscope (SEM), X-ray diffraction (XRD) and energy dispersive spectroscopy (EDS) were employed to characterize. The effect of DIOP on the apatite forming ability was investigated after immersion in SBF. The attachment and proliferation of the MG-63 cells on the porous scaffolds were evaluated to assess the cytocompatibility.
2 Experimental
2.1 Materials
PGA powders (Mw = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) with mean particle size of about 40 μm were provided by Shenzhen Polymtek Biomaterial Co., Ltd (Shenzhen, China). DIOP powders consisting of 55 wt% SiO2, 24 wt% CaO and 18 wt% MgO with mean particle size of about 200 nm were purchased from Kunshan Chinese Technology New Materials (Kunshan, China). Human osteoblast-like MG-63 cells line used for cell culture were provided by American Type Culture Collection (ATCC, Rockville, MD). Phosphate buffer solution (PBS) was purchased from Sigma-Aldrich (Beijing, China). Fetal bovine serum (FBS) and Dulbecco's modified Eagle's medium (DMEM) were both purchased from Life Technologies (Gibco®, Carlsbad, CA, USA).
000 Da) with mean particle size of about 40 μm were provided by Shenzhen Polymtek Biomaterial Co., Ltd (Shenzhen, China). DIOP powders consisting of 55 wt% SiO2, 24 wt% CaO and 18 wt% MgO with mean particle size of about 200 nm were purchased from Kunshan Chinese Technology New Materials (Kunshan, China). Human osteoblast-like MG-63 cells line used for cell culture were provided by American Type Culture Collection (ATCC, Rockville, MD). Phosphate buffer solution (PBS) was purchased from Sigma-Aldrich (Beijing, China). Fetal bovine serum (FBS) and Dulbecco's modified Eagle's medium (DMEM) were both purchased from Life Technologies (Gibco®, Carlsbad, CA, USA).
2.2 Fabrication of porous scaffolds
A schematic diagram for the experimental procedure to fabricate the porous scaffolds was shown in Fig. 1. That is: the PGA powders were added to 20 ml of ethyl alcohol and ultrasonically dispersed for 10 min. Then DIOP powders were added to the solution, and the solution was ultrasonically dispersed for additional 10 min. The suspension was filtered immediately following ultrasonic dispersion. The composite powders were dried overnight in an electrothermal blowing dry box (101-00S, China), followed by mechanical mixing for 30 min. Five types of PGA/DIOP composite powders containing 2.5, 5, 7.5, 10 and 12.5 wt% of DIOP were obtained. The porous scaffolds were prepared using the SLS system, which was detailedly described in our previous study.25 Briefly, the computer-aided design (CAD) model of the porous scaffold was first converted to a stereolithography (STL) format then transferred to the sintering machine for scaffold fabrication. And then the sintering paths (layer by layer) were planned. The sintering-process parameters, such as scanning speed, laser power, layer thickness and spot diameter, were 200 mm min−1, 2.5 W, 0.1–0.2 mm and 0.8 mm, respectively. After fabrication, the scaffold was carefully retrieved from the powder bed and cleaned by blowing compressed air to remove the excess powders. | ||
| Fig. 1 The schematic diagram of the experimental procedures to fabricate PGA porous scaffolds. | ||
2.3 Characterization
Microstructural observations of the raw powders and porous scaffolds were performed using a FEI Quanta-200 (FEI Co., USA) at 20 kV. For SEM observation, specimes were coated with platinum for 200 s under a current of 20 mA using a JFC-1600 sputter fine coater (JEOL, Ltd., Japan). XRD analysis was performed to detect the phase composition of the scaffolds on a Bruker D8 Advance Diffractometer (German Bruker Co., German) using nickel filtered Cu-Kα radiation and operating at 40 kV/40 mA. The specimes were scanned over a 2θ range of 10–50° with 0.02° increment at a rate of 8° min−1.The compressive mechanical properties of the scaffold specimes (15 × 15 × 12 mm3) were determined using a WD-D1 universal testing machine (Shanghai Zhuoji instruments Co. LTD, China). The specimens were compressed between platens, and the crosshead speed was 0.5 mm min−1. Compressive modulus was obtained from the stress–strain curve by measuring the slope of the initial linear portion. Six identical specimens of each formulation were analyzed for scaffolds' compressive properties. Data in this study were determined as mean value ± standard deviation (SD). Statistical difference was analyzed using Origin 8.0 (OriginLab Co., USA) and statistically significance and remarkably significance were defined as p value of <0.05 and <0.01, respectively.
2.4 Apatite forming ability in SBF
The apatite forming ability was assessed by soaking the scaffold specimens (15 × 15 × 12 mm3) in SBF solution. The inorganic ion concentrations in the SBF were: 142.0 mM, 5.0 mM, 2.5 mM, 1.5 mM, 148.8 mM, 4.2 mM, 1.0 mM and 0.5 mM for Na+, K+, Ca2+, Mg2+, Cl−, HCO3−, HPO42− and SO42−, respectively. Tris(hydroxymethyl)-aminomethane and 1 M HCl were used to maintain pH of the solution at 7.4. The scaffold specimens were immersed in the SBF at a concentration of 10 mm2 mL−1 with a volume of 50 mL, and the SBF was renewed every day. The solutions with the specimens were put in an incubator and maintained at 37 °C. After soaking for 7 days the specimens were extracted from the solution, carefully washed with deionized water, and dried at 37 °C in the electrothermal blowing dry box for 2 h. The microstructure and elemental composition of the deposited phases on specimen surface after soaking was characterized using SEM coupled with EDS. Three points of each specimen were measured to calculate the Ca/P ratio. The date were experssed as means ± SD and a p-value <0.05 was considered statistically significant.2.5 Degradation in PBS
In vitro degradation evaluation of the scaffold was performed in PBS solution at pH 7.4 and 37 °C for 1 day, 3, 7 and 14 days. The scaffold specimens were weighed and then placed in polystyrene bottles containing 100 ml PBS solution. The pH value of the PBS solution was measured by a pH meter at each interval. At the selected time points, six specimens of each scaffold were removed from the buffer, rinsed with deionized distilled water and dried in the dry box at 37 °C for 24 h. Degradation rate of each scaffold specimen was measured in terms of weight loss at different time. The weight loss (WL) was calculated as follows:| WL(%) = (W0 − Wt)/W0 × 100 |
2.6 In vitro cell culture
The cytocompatibility tests of the scaffolds was performed using MG-63 cell. The line was resuspended in DMEM supplemented with 10% FBS and 5% penicillin/streptomycin antibiotics. Before cell seeding, the scaffold specimens (15 × 15 × 12 mm3) were cleaned using 70% ethanol in an orbital shaker, and sterilized for 30 min under UV light. The scaffolds were then immersed in culture medium overnight before cell seeding. MG-63 cells were seeded onto the top of each specimen at a density of 2 × 105 cells per cm2 and cultured in 24-well plates for 4 h, 1, 3 and 5 days. At the specified time point, the specimens were removed and rinsed three times with PBS, fixed with 2.5% glutaraldehyde, dehydrated in varying concentrations of ethanol (50%, 60%, 70%, 80%, 90%, and 100% v/v) for 20 min, dried in the electrothermal blowing dry box 12 h. The specimens were sputtered with platinum and observed by SEM.2.7 Cell proliferation and differentiation
Cell proliferation of the pure PGA-10 wt% DIOP and the PGA scaffold was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. The cell-scaffold constructs were placed in culture medium containing MTT and incubated in a humidified atmosphere at 37 °C for 4 h. And then the optical density was recorded at 570 nm using an enzyme immunosorbent assay reader. MG-63 cell differentiation was analyzed after different culture time by measuring the alkaline phosphatase (ALP) activity. The adherent cells were removed, washed in PBS three times, mixed with cell lysis buffer containing 0.1% Triton X, and incubated at 4 °C for 2 h. The supernatant was tested for ALP activity and absorbance was read at 405 nm. The ALP activity of the MG-63 cells was assayed using a Laboassay™ ALP kit (Wako Pure Chemicals, Japan) according to the manufacturer's instructions.3 Results and discussion
The top-view and side-view SEM micrographs of the porous scaffold were shown in Fig. 2. It could be seen that the scaffold had a well-controlled pore structure with completely interconnected porous system. The interconnecting channels of the porous scaffolds were very important for cellular attachment and growth, vascular and tissue ingrowth, and nutrient delivery and waste expulsion.26 The average size of the pores and struts were about 1100 μm and 1050 μm, respectively. Previous studies have reported that macro- and micro-structures of scaffolds have great contribute to the development of specific biological functions in tissues and provide appropriate nutritional conditions and spatial organization for cell growth.4,27–29 For example, Jonnalagadda et al.4 pointed that the optimal pore size of scaffolds was 100–500 μm for tissue engineering and 70–120 μm for chondrocyte growth. Wei et al.27 showed that the macropore diameter of 400–500 μm is beneficial in facilitating cell infiltration and bone ingrowth and micropore of 10–20 μm plays an important role in nutrient diffusion. While, it has been reported that pore size greater than 300 μm is essential for vascularisation of scaffolds and bone ingrowth.30–33 Roy et al.33 found that there was no significant difference in bone growth for 500 and 1600 μm pore size for PLGA scaffolds. Hollister et al.34 found that significant bone growth on designed scaffolds for all pores (400 and 1200 μm), with no statistical difference between pore size. | ||
| Fig. 2 Top-view (a, c and d) and side-view (b, e and f) SEM micrographs for the porous scaffold. | ||
The SEM images of the raw PGA and DIOP powders were presented in Fig. 3a and b, respectively. The raw PGA powders had irregular shapes or a shell-shape, which varied in particle size from 10 μm to 70 μm (Fig. 3a). The DIOP powders had a spherical shape with uniformed particle size (Fig. 3b). To study the dispersion state of DIOP powders in the PGA matrix, the surface morphologies of scaffolds with different DIOP content were shown in Fig. 3c–h. The pure PGA scaffold exhibited a smooth surface (Fig. 3c). A uniform dispersion of DIOP particles in PGA matrix was observed for the scaffolds with 2.5, 5, 7.5 and 10 wt% DIOP and the agglomeration of DIOP particles was negligible (Fig. 3d–g). In the scaffold containing 12.5 wt% DIOP (Fig. 3h), a significantly agglomeration was observed and the particles were non-uniformly distributed in the PGA matrix.
 | ||
| Fig. 3 (a) The raw PGA powders, (b) the raw DIOP powders, (c) the pure PGA scaffold, (d) the PGA-2.5 wt% DIOP scaffold, (e) the PGA-5 wt% DIOP scaffold, (f) the PGA-7.5 wt% DIOP scaffold, (g) the PGA-10 wt% DIOP scaffold and (h) the PGA-12.5 wt% DIOP scaffold. | ||
The XRD patterns of the raw PGA and DIOP powders, the pure PGA scaffold and the DIOP reinforced PGA scaffolds containing 2.5, 5, 7.5, 10 and 12.5 of DIOP were depicted in Fig. 4. The raw PGA powders contained two strong reflection peaks at the angles (2θ) of about 22.2 and 28.9 and one weak reflection peak at the angles (2θ) of about 35.8, corresponding to the (110), (020) and (121) planes of the crystal structure, respectively.35 The peaks of DIOP could be seen at 2θ = 26.8, 27.9, 29.8, 30.8 35.0 and 35.7 according to the JCPDS file number 01-075-1092.36 The pure PGA scaffold pattern contained only the PGA characteristic peaks. Comparing the XRD pattern of the pure PGA scaffold with DIOP modified PGA scaffolds, it could be seen that the strong reflection peaks observed from PGA were also presented in the scaffolds. The reflection peak at the angles (2θ) of about 26.8 appeared when the DIOP content was above 2.5 wt%. Increasing the DIOP to 12.5 wt% resulted in more intensive DIOP peaks.
 | ||
| Fig. 4 XRD patterns of (a) the raw PGA powders, (b) the raw DIOP powders, (c) the pure PGA scaffold, (d) the PGA-2.5 wt% DIOP scaffold, (e) the PGA-5 wt% DIOP scaffold, (f) the PGA-7.5 wt% DIOP scaffold, (g) the PGA-10 wt% DIOP scaffold, (h) the PGA-12.5 wt% DIOP scaffold. | ||
The scaffolds' compressive strength and modulus as a function of the DIOP content were shown in Fig. 5. The scaffolds' compressive strength increased from 10.57 MPa up to 28.67 MPa (increased by 171.2%) and compressive modulus increased from 2.36 GPa up to 3.45 GPa (increased by 46.2%) with the increase in DIOP concentration up to 10 wt%. Ghorbanian et al.37 prepared diopside/silk fibroin composite scaffold using a freeze-drying method and found that the addition of diopside in the silk fibroin matrix abtained a 20% reinforcement in strength. The compressive strength of the scaffold was in the range of that of human cancellous bone (0.1–16 MPa) but much lower than that of cortical bone (130–180 MPa).38,39 The DIOP addition significantly improved the mechanical properties of the PGA scaffolds (p < 0.05). The DIOP powders acted as stiff filler in PGA matrix and therefore enhanced scaffolds' mechanical properties. While further increase of DIOP content beyond 12.5 wt%, the agglomeration of the DIOP powders occurred, which resulted in the decrease of the mechanical properties. It was well known that the agglomeration of stiff filler within polymer matrix could deteriorate the mechanical properties of the composites.40–42 So, the optimal DIOP content was 10 wt% at the highest mechanical properties.
 | ||
| Fig. 5 Mean values of the scaffolds' compressive strength and modulus as a function of the DIOP content. Error bars represent means ± SD for n = 6 (*p < 0.05, **p < 0.01). | ||
Besides mechanical properties, bioactivity is another important characteristic of porous scaffolds for bone tissue application. The scaffolds' in vitro bioactivity with different DIOP content was evaluated by examining the apatite formation on their surfaces in SBF. The surface morphology and EDS profiles of the scaffolds after SBF immersion were shown in Fig. 6. No sign of particles deposition was found on pure PGA scaffold surface (Fig. 6a). EDS analysis revealed the presence of C, O, Pt, Na and Cl element, but no Ca and P element. It indicated that apatite could not precipitate on the pure PGA scaffold surface. Cl and Na elements were detected, which likely came from SBF itself.43 Only a small number of deposits were observed on the PGA-2.5 wt% DIOP scaffold surface (Fig. 6b). The number of particles deposition increased with the increase of DIOP content. A layer of particles formed on the PGA-12.5 wt% DIOP scaffold (Fig. 6f). The newly formed layer was composed of nanocrystals with worm-like morphology. EDS detected Ca and P characteristic peaks, and the Ca/P ratio was 1.592 ± 0.011, similar to 1.67 Ca/P ratio of hydroxyapatite.44,45 The Ca/P ratio of the PGA-12.5 wt% DIOP scaffold was significantly higher than that for other scaffolds (p < 0.05). It indicated that the formation of bone-like apatite on the scaffold surface. These results showed that addition of DIOP particles into the PGA matrix stimulated and accelerated apatite formation, and thus enhanced the bioactive behavior of scaffold.
 | ||
| Fig. 6 SEM micrographs and EDS profiles for (a) the pure PGA scaffold, (b) the PGA-2.5 wt% DIOP scaffold, (c) the PGA-5 wt% DIOP scaffold, (d) the PGA-7.5 wt% DIOP scaffold, (e) the PGA-10 wt% DIOP scaffold and (f) the PGA-12.5 wt% DIOP scaffold soaked in SBF for 7 days. | ||
In vitro degradation behavior of scaffold is an important factor in bone tissue regeneration. The weight loss of the pure PGA and PGA-10 wt% DIOP scaffold as a function of immersion time was shown in Fig. 7a. Weight loss of the pure PGA scaffold was higher than that of the PGA-10 wt% DIOP scaffold and increased gradually as immersion time prolonged. It indicated that the addition of DIOP in the PGA matrix could decreased the scaffolds' degradation rate. The pH change of the PBS solution during in vitro degradation as a function of the immersion time was shown in Fig. 7b. The pH of the solution for the pure PGA scaffold decreased from the initial value of 7.4, which was due to the released acidic products from PGA degradation. The pH of the PGA-10 wt% DIOP scaffold did not change significantly, which might be correlated to the release of alkaline ions from DIOP, such as Ca2+, Mg2+ and Si4+ that neutralizes the acidification of the medium due to PGA degradation. Previous studies also reported this similar buffering effects in bioglass, forsterite and wollastonite.46–48 There were significant differences in weight loss and solution pH after immersion in SBF between the PGA-10 wt% DIOP and the pure PGA scaffold (p < 0.05).
 | ||
| Fig. 7 (a) Weight loss in the pure PGA and PGA-10 wt% DIOP scaffold after soaking in PBS, (b) variation of pH value of the PBS solution during in vitro degradation. Error bars represent means ± SD for n = 6 (*p < 0.05, **p < 0.01). | ||
An ideal scaffold should be bioactive, which could bond to living bone through a bone-like apatite layer.49 Therefore, the apatite layer is very important for the bone forming.50,51 In the present study, the PGA-DIOP scaffolds possessed good apatite formation ability. Ionic exchange between DIOP and SBF solution might take place during incubation, which would drive silanol-like group (Si–OH) formation and therefore induce the apatite nucleation.52,53 A bone-like apatite layer would grow from these apatite nuclei by expensing of P ions.54,55 Biodegradable scaffolds provide the initial structure and stability for tissue formation but degrade as the tissue forms, providing room for matrix deposition and tissue growth.56 Our results showed that the DIOP modification of PGA scaffold could adjust the degradation rate and pH value of the environment. Previous studies have proved that the degradation of Ca-, Mg- and Si-containing bioceramics could generate alkaline ions and neutralize the acids products of polymers.57,58
Osteoblast-like cells attachment and spreading is a prerequisite for their growth, differentiation and migration.59 MG-63 cells attachment and proliferation on the pure PGA and PGA-10 wt% DIOP scaffolds were shown in Fig. 8. The cells could adhere and spread on all scaffolds, and increased in density with increasing incubation time. The cells adhered on pure PGA scaffold, but did not spread well (Fig. 8a). They attached well and spread out with thin filopodia at the periphery (Fig. 8b). As the culture time increased, more cells stretched and proliferated on the scaffolds surface (Fig. 8c–e). The MG-63 cells attachment and spreading on the PGA-DIOP scaffolds was significantly improved than that on the pure PGA scaffold (Fig. 8f–j). They had a well-spread and flattened morphology. After 5 days of culture, the cells coalesced to form a flat layer and covered the whole scaffolds surface (Fig. 8i and j). The results indicated that MG-63 cells on the DIOP modified PGA scaffolds exhibited a high degree of cell attachment, growth as well as differentiation compared to those on the pure PGA scaffold.
 | ||
| Fig. 8 SEM micrographs of MG-63 cells cultured on the pure PGA scaffold for (a) 6 hours, (b) 1 day, (c) 3 days, (d) 5 days, (e) 7 days, and the PGA-10 wt% DIOP scaffold for (f) 6 hours, (g) 1 day, (h) 3 days, (i) 5 days, (j) 7 days. | ||
The proliferation of MG-63 cells on the PGA-10 wt% DIOP and the pure PGA scaffold was assessed by MTT assay (Fig. 9a). The cell number on the PGA-10 wt% DIOP scaffold was higher than that on the pure PGA scaffold after 1 day of culture. The cell number on both scaffolds increased and the difference in cell number on the PGA-10 wt% DIOP scaffold and the PGA scaffold also increased as the culture time prolonged. The cell number on the PGA-10 wt% DIOP scaffold were significantly higher than that on the pure PGA scaffold after 3 days of culture (p < 0.05). Cell differentiation was evaluated in terms of the ALP activities of MG-63 cells cultured on the PGA-10 wt% DIOP scaffold at 6 h, 1, 3, 5 and 7 days (Fig. 9b–f). The ALP activity of the MG-63 cells was expressed at lower levels at 6 h (Fig. 4b) and 1 day (Fig. 9c) on the scaffolds. While, the level of ALP activity increased substantially on the scaffolds with incubation period prolonging (Fig. 9d–f).
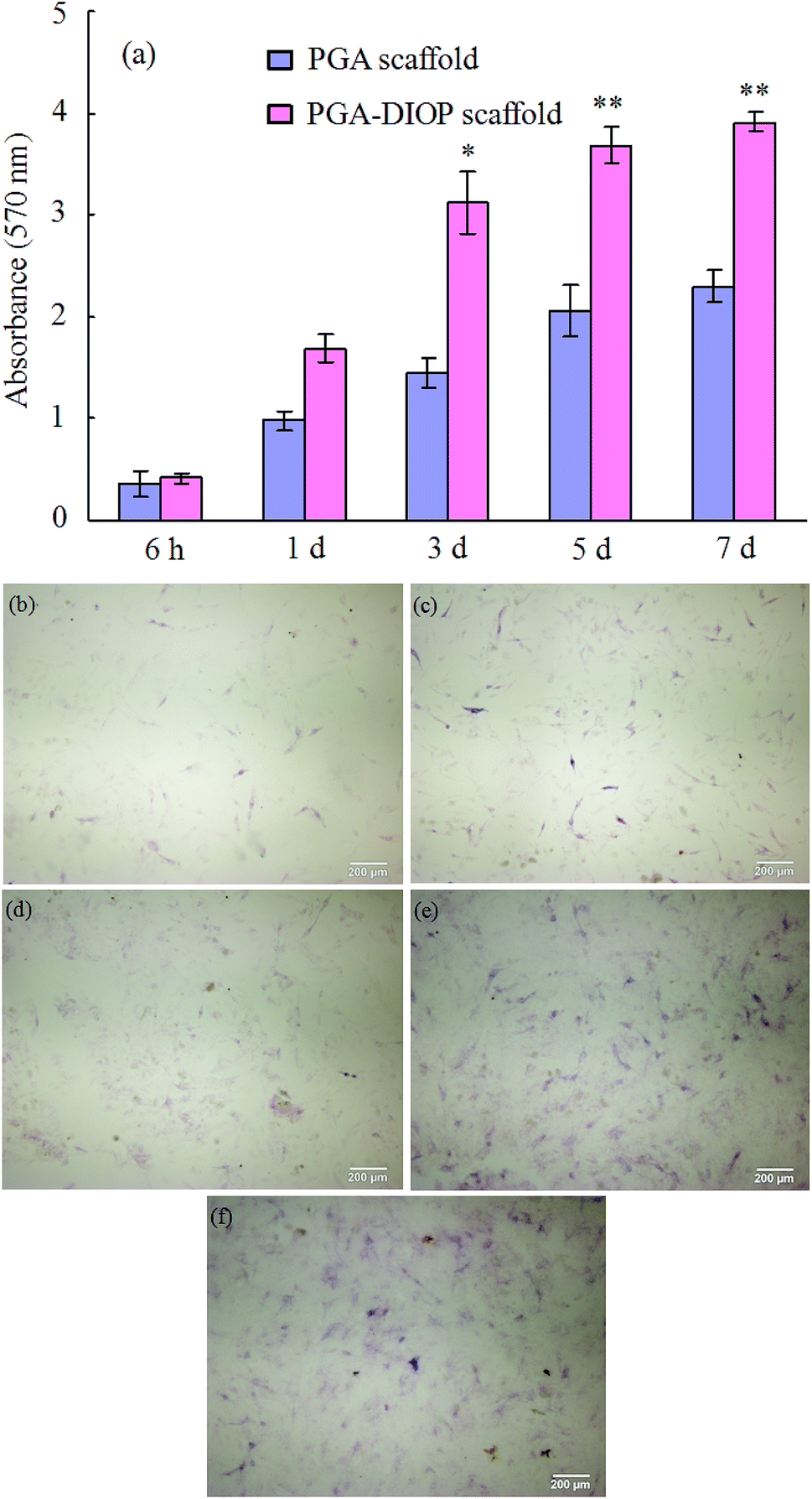 | ||
| Fig. 9 Proliferation (a) of MG-63 cells on the PGA-10 wt% DIOP and the pure PGA scaffold after culture for different time and ALP (b–f) activity of MG-63 cells on the PGA-10 wt% DIOP scaffold after culture for different time. Data presented as mean ± SD, n = 6. *p < 0.05, **p < 0.01. | ||
The improvement of cell biocompatibility was the result of several factors working together at the same time. For instance, DIOP could stimulate the bone-like apatite deposition, which provided a suitable substrate for cell attachment and differentiation.60,61 In addition, the degradation of DIOP and the release of Ca, Si and Mg ions might neutralized the acidic degradation products from PGA and resulted in the stability of pH value. Previous studies have demonstrated that low or high pH might inhibit cells behavior.62–64 Moreover, the Si–OH group formation due to the addition of DIOP led to enhanced cells adhesion and proliferation.65,66
4 Conclusions
Porous PGA scaffolds modified with DIOP were fabricated using SLS and the influences of DIOP content on the mechanical and biological properties were evaluated. The results showed that the DIOP could significantly improve the compressive strength and modulus of the scaffolds. In addition, the in vitro studies in SBF showed that the DIOP could improve the apatite formation ability of the scaffolds and this ability increased with increasing DIOP content in the PGA matrix. The results of the cell culture study revealed that the scaffolds supported the MG-63 cells adhesion and proliferation with enhanced spreading ability more than the pure PGA scaffold, indicating that the DIOP modified scaffolds have great potential for bone tissue regeneration.Acknowledgements
This work was supported by the following funds: (1) The Natural Science Foundation of China (51222506, 81372366, 81472058); (2) High Technology Research and Development Program of China (2015AA033503); (3) Overseas, Hong Kong & Macao Scholars Collaborated Researching Fund of National Natural Science Foundation of China (81428018); (4) Hunan Provincial Natural Science Foundation of China (14JJ1006); (5) Shenzhen Strategic Emerging Industrial Development Funds (JCYJ20130401160614372); (6) The faculty research grant of Central South University (2013JSJJ011, 2013JSJJ046); (7) State Key Laboratory of New Ceramic and Fine Processing Tsinghua University (KF201413); (8) The Open-End Fund for the Valuable and Precision Instruments of Central South University; (9) The Fundamental Research Funds for the Central Universities of Central South University.Notes and references
- G. Bernardini, F. Chellini, B. Frediani, A. Spreafico and A. Santucci, J. Biosci., 2015, 40, 1 CrossRef.
- H. Cao and N. Kuboyama, Bone, 2010, 46, 386 CrossRef CAS PubMed.
- H. Xu, D. Han, J. S. Dong, G. X. Shen, G. Chai, Z. Y. Yu, W. J. Lang and S. T. Ai, Int. J. Med. Robot. Comput. Assist. Surg., 2010, 6, 66 Search PubMed.
- J. B. Jonnalagadda, I. V. Rivero and J. S. Dertien, J. Biomater. Sci., Polym. Ed., 2015, 26, 401 CrossRef CAS PubMed.
- R. M. Aghdam, S. Najarian, S. Shakhesi, S. Khanlari, K. Shaabani and S. Sharifi, J. Appl. Polym. Sci., 2012, 124, 123 CrossRef CAS PubMed.
- Q. Zhao, S. Wang, J. Tian, L. Wang, S. Dong, T. Xia and Z. Wu, J. Mater. Sci.: Mater. Med., 2013, 24, 793 CrossRef CAS PubMed.
- C. Wu, Y. Ramaswamy, Y. Zhu, R. Zheng, R. Appleyard, A. Howard and H. Zreiqat, Biomaterials, 2009, 30, 2199 CrossRef CAS PubMed.
- J. M. Pachence and J. Kohn, Princ. Tissue Eng., 2000, 3, 323 Search PubMed.
- T. Miyai, A. Ito, G. Tamazawa, T. Matsuno, Y. Sogo, C. Nakamura, A. Yamazaki and T. Satoh, Biomaterials, 2008, 29, 350 CrossRef CAS PubMed.
- Z. Dong, Y. Li and Q. Zou, Appl. Surf. Sci., 2009, 255, 6087 CrossRef CAS PubMed.
- M. Sadat-Shojai, M. T. Khorasani, A. Jamshidi and S. Irani, Mater. Sci. Eng., C, 2013, 33, 2776 CrossRef CAS PubMed.
- M. Selvakumar, S. K. Jaganathan, G. B. Nando and S. Chattopadhyay, J. Biomed. Nanotechnol., 2015, 11, 291 CrossRef CAS PubMed.
- A. J. Harmata, C. L. Ward, K. J. Zienkiewicz, J. C. Wenke and S. A. Guelcher, J. Mater. Res., 2014, 29, 2398 CrossRef CAS PubMed.
- B. Lei, K. H. Shin, D. Y. Noh, I. H. Jo, Y. H. Koh, H. E. Kim and S. E. Kim, Mater. Sci. Eng., C, 2013, 33, 1102 CrossRef CAS PubMed.
- B. Lei, K. H. Shin, D. Y. Noh, Y. H. Koh, W. Y. Choi and H. E. Kim, J. Biomed. Mater. Res., Part B, 2012, 100, 967 CrossRef PubMed.
- Y. Zhang, S. Li and C. Wu, J. Biomed. Mater. Res., Part A, 2014, 102, 105 CrossRef PubMed.
- E. Ercenk, J. Non-Cryst. Solids, 2014, 387, 101 CrossRef CAS PubMed.
- J. P. Kumar, L. Lakshmi, V. Jyothsna, D. R. Balaji, S. Saravanan, A. Moorthi and N. Selvamurugan, J. Biomed. Nanotechnol., 2014, 10, 970 CrossRef CAS PubMed.
- C. Wu and J. Chang, Biomed. Mater., 2013, 8, 031001 Search PubMed.
- T. Nonami and S. Tsutsumi, J. Mater. Sci.: Mater. Med., 1999, 10, 475 CrossRef CAS.
- A. Harabi and S. Zouai, Int. J. Appl. Ceram. Technol., 2014, 11, 31 CrossRef CAS PubMed.
- S. Nakajima, Y. Harada, Y. Kurihara, T. Wakatsuki and H. Noma, Dent. Sci. Rep., 1989, 89, 1709 CAS.
- A. Nadernezhad, B. Torabinejad, M. Hafezi, M. Baghban-Eslaminejad, F. Bagheri and F. Najafi, J. Adv. Mater. Process., 2014, 2, 13 Search PubMed.
- C. Wu, Y. Ramaswamy and H. Zreiqat, Acta Biomater., 2010, 6, 2237 CrossRef CAS PubMed.
- C. Shuai, P. Feng, L. Zhang, C. Gao, H. Hu, S. Peng and A. Min, Sci. Technol. Adv. Mater., 2013, 14, 055002 CrossRef.
- L. C. Gerhardt and A. R. Boccaccini, Materials, 2010, 3, 3867 CrossRef CAS PubMed.
- J. Wei, X. Wu, C. Liu, J. Jia, S. Heo, S. Kim, Y. Hyun and J. Shin, J. Am. Ceram. Soc., 2009, 92, 1017 CrossRef CAS PubMed.
- S. Yang, K. F. Leong, Z. Du and C. K. Chua, Tissue Eng., 2001, 7, 679 CrossRef CAS PubMed.
- A. R. Amini, D. J. Adams, C. T. Laurencin and S. P. Nukavarapu, Tissue Eng., Part A, 2012, 18, 1376 CrossRef CAS PubMed.
- C. M. Murphy, M. G. Haugh and F. J. O'Brien, Biomaterials, 2010, 31, 461 CrossRef CAS PubMed.
- K. C. R. Kolan, M. C. Leu, G. E. Hilmas, R. F. Brown and M. Velez, Biofabrication, 2011, 3, 025004 CrossRef PubMed.
- M. Dadsetan, T. E. Hefferan, J. P. Szatkowski, P. K. Mishra, S. I. Macura, L. Lu and M. J. Yaszemski, Biomaterials, 2008, 29, 2193 CrossRef CAS PubMed.
- T. D. Roy, J. L. Simon, J. L. Ricci, E. D. Rekow, V. P. Thompson and J. R. Parsons, J. Biomed. Mater. Res., Part A, 2003, 66, 283 CrossRef PubMed.
- S. J. Hollister, C. Y. Lin, E. Saito, R. D. Schek, J. M. Taboas, J. M. Williams, B. Partee, C. L. Flanagan, A. Diggs, E. N. Wilke, G. H. Van Lenthe, R. Müller, T. Wirtz, S. Das, S. E. Feinberg and P. H. Krebsbach, Orthod. Craniofac. Res., 2005, 5, 162 CrossRef PubMed.
- X. H. Zong, Z. G. Wang, B. S. Hsiao, B. Chu, J. J. Zhou, D. D. Jamiolkowski, E. Muse and E. Dormier, Macromolecules, 1999, 32, 8107 CrossRef CAS.
- K. Otto, W. Wisniewski and C. Rüssel, CrystEngComm, 2013, 15, 6381 RSC.
- L. Ghorbanian, R. Emadi, S. M. Razavi, H. Shin and A. Teimouri, Int. J. Biol. Macromol., 2013, 58, 275 CrossRef CAS PubMed.
- P. Feng, P. Wei, C. Shuai and S. Peng, PLoS One, 2014, 9, e87755 Search PubMed.
- X. P. Fan, B. Feng, Y. L. Di, J. X. Wang, X. Lu and J. Weng, Powder Metall. Met. Ceram., 2012, 51, 372 CrossRef CAS.
- J. Y. Liu, L. Reni, Q. Wei, J. L. Wu, S. Liu, Y. J. Wang and G. Y. Li, eXPRESS Polym. Lett., 2011, 5, 742 CrossRef CAS.
- M. Diba, M. Kharaziha, M. H. Fathi, M. Gholipourmalekabadi and A. Samadikuchaksaraei, Compos. Sci. Technol., 2012, 72, 716 CrossRef CAS PubMed.
- S. K. Misra, D. Mohn, T. J. Brunner, W. J. Stark, S. E. Philip, I. Roy, V. Salih, J. C. Knowles and A. R. Boccaccini, Biomaterials, 2008, 29, 1750 CrossRef CAS PubMed.
- P. M. Vilarinho, N. Barroca, S. Zlotnik, P. Félix and M. H. Fernandes, Mater. Sci. Eng., C, 2014, 39, 395 CrossRef CAS PubMed.
- M. Peter, N. S. Binulal, S. V. Nair, N. Selvamurugan, H. Tamura and R. Jayakumar, Chem. Eng. J., 2010, 158, 353 CrossRef CAS PubMed.
- Y. J. Ye, P. Y. Wang, Y. P. Li and D. C. Yin, J. Mater. Sci.: Mater. Med., 2015, 26, 1 CAS.
- S. I. Roohani-Esfahani, S. Nouri-Khorasani, Z. F. Lu, R. C. Appleyard and H. Zreiqat, Acta Biomater., 2011, 7, 1307 CrossRef CAS PubMed.
- M. Diba, M. Kharaziha, M. H. Fathi, M. Gholipourmalekabadi and A. Samadikuchaksaraei, Compos. Sci. Technol., 2012, 72, 716 CrossRef CAS PubMed.
- J. Wei, F. Chen, J. W. Shin, H. Hong, C. Dai, J. Su and C. Liu, Biomaterials, 2009, 30, 1080 CrossRef CAS PubMed.
- Y. Zhu and S. Kaskel, Microporous Mesoporous Mater., 2009, 118, 176 CrossRef CAS PubMed.
- C. Wu, J. Chang, W. Zhai and S. Ni, J. Mater. Sci.: Mater. Med., 2007, 18, 857 CrossRef CAS PubMed.
- A. J. Salinas and M. Vallet-Regí, RSC Adv., 2013, 3, 11116 RSC.
- M. Karanjai, R. Sundaresan, T. R. R. Mohan and B. P. Kashyap, Mater. Sci. Eng., C, 2008, 28, 1401 CrossRef CAS PubMed.
- C. Ohtsuki, M. Kamitakahara and T. Miyazaki, J. R. Soc., Interface, 2009, 6, S349 CrossRef CAS PubMed.
- S. G. Caridade, E. G. Merino, N. M. Alves and J. F. Mano, Macromol. Biosci., 2012, 12, 1106 CrossRef CAS PubMed.
- G. Liu, C. Wu, W. Fan, X. Miao, D. C. Sin, R. Crawford and Y. Xiao, J. Biomed. Mater. Res., Part A, 2011, 96, 360 CrossRef PubMed.
- W. S. Khan, F. Rayan, B. S. Dhinsa and D. Marsh, Stem Cells Int., 2012, 22, 12288 Search PubMed.
- E. Bernardo, J. F. Carlotti, P. M. Dias, L. Fiocco, P. Colombo, L. Treccani, U. Hess and K. Rezwan, Ceram. Int., 2014, 40, 1029 CrossRef CAS PubMed.
- L. Chen, D. Zhai, C. Wu and J. Chang, Ceram. Int., 2014, 40, 12765 CrossRef CAS PubMed.
- T. V. Chirila, Z. Barnard, D. G. Harkin, I. R. Schwab and L. W. Hirst, Tissue Eng., Part A, 2008, 14, 1203 CrossRef CAS.
- P. Feng, C. Gao, C. Shuai and S. Peng, RSC Adv., 2015, 5, 3498 RSC.
- S. Bose, M. Roy and A. Bandyopadhyay, Trends Biotechnol., 2012, 30, 546 CrossRef CAS PubMed.
- W. Huang, W. Choi, Y. Chen, Q. Zhang, H. Deng, W. He and Y. Shi, Cell Res., 2013, 23, 724 CrossRef CAS PubMed.
- Z. T. Cusumano and M. G. Caparon, J. Bacteriol., 2015, 197, 1288 CrossRef CAS PubMed.
- O. Bretcanu, A. R. Boccaccini and V. Salih, J. Mater. Sci., 2012, 47, 5661 CrossRef CAS.
- H. Zhu, J. Shen, X. Feng, H. Zhang, Y. Guo and J. Chen, Mater. Sci. Eng., C, 2010, 30, 132 CrossRef CAS PubMed.
- S. Chen, A. Osaka, T. Ikoma, H. Morita, J. Li, M. Takeguchi and N. Hanagata, J. Mater. Chem., 2011, 21, 10942 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |