
A series of multidimensional MOFs incorporating a new N-heterocyclic building block: 5,5′-di(pyridin-4-yl)-3,3′-bi(1,2,4-triazole)†
Peng-Fei Yaoa,
Hai-Ye Lia,
Fu-Ping Huang*a,
Qing Yua,
Xao-Huan Qina,
Jie Tanga,
Ye Taoa and
He-Dong Bian*b
aKey Laboratory for the Chemistry and Molecular Engineering of Medicinal Resources (Ministry of Education of China), School of Chemistry and Pharmacy, Guangxi Normal University, Guilin, 541004, P. R. China. E-mail: huangfp2010@163.com
bSchool of Chemistry and Chemical Engineering, Guangxi University for Nationalities, Key Laboratory of Chemistry and Engineering of Forest Products, Nanning, 530008, P. R. China. E-mail: gxunchem@163.com
First published on 18th May 2015
Abstract
By using a new N-heterocyclic building block, 5,5′-di(pyridin-4-yl)-3,3′-bi(1,2,4-triazole) (4,4′-H2dbpt), six novel coordination polymers with diversiform connectivity from one- to three-dimensional were successfully constructed. By regulating the different auxiliary ligands, central metal ions, and some other synthetic conditions, 4,4′-H2dbpt adopted various coordination modes. Consequently, 1 adopts a 2D (3,6)-topology, with the (43)3(46.66.83)2 Schläfli symbol. 2 shows a 3D 8-connected topology with a (36.418.53.6) Schläfli symbol. 3 and 4, which are isostructural, both have a 2D 4-connected topology, with a (44.62) Schläfli symbol. 5 has a complex 3D porous architecture with a 1D solvent-filled channel. 6 reveals a 1D helical chain extended along a 4-fold screw axis. These results indicate that 4,4′-H2dbpt is an excellent multi-connection linker from which we can construct MOFs with interesting structures and properties.
Introduction
The design and synthesis of coordination polymers, especially metal–organic frameworks (MOFs) has attracted considerable interest in the realm of supramolecular chemistry and crystal engineering, owing not only to their appealing structural and topological novelty but also to their tremendous potential applications in gas storage and separation,1 electrical conduction,2 and as luminescence materials,3 molecular magnets,4 and heterogeneous catalysts.5 Thus, a series of studies in this field have mainly focused on the design, preparation, and structure–property relationships.6 The structures of coordination polymers are determined by several factors, including the coordination geometry of the central metal ion, solvents,7 ligand structure, metal-ligand ratio,8 counterions,9 pH,10 and temperature.11 Among the reported studies, much effort has been focused on the rational design and controlled synthesis of coordination polymers using multidentate ligands such as polycarboxylate and N-heterocyclic ligands. The N-heterocyclic ligands are good molecular building blocks and co-ligands for constructing MOFs with interesting structures and properties, which have been widely reported by Yaghi,12 Snurr,13 Chen,14 Du,15 Tong,16 and Zhang.17Our groups have systematically explored the coordination assemblies on the basis of some polycarboxylate ligands that cooperate with some angular N-heterocyclic-like ligands: 1H-3,5-bis(4-pyridyl)-1,2,4-triazole (4,4′-bpt), 1H-3-(3-pyridyl)-5-(4-pyridyl)-1,2,4-triazole (3,4′-bpt), 1H-3,5-bis(3-pyridyl)-1,2,4-triazole (3,3′-bpt), 4-amino-3,5-bis(4-pyridyl)-1,2,4-triazole (4,4′-abpt), and 1,4-bis(5-(4-pyridyl)-1H-1,2,4-triazol-3-yl)benzene (bptb).18 In most cases, the angular N-heterocyclic-like ligands act as 2-connected linkers with different distortion angles and directions. Relying on the versatile coordination modes of the polycarboxylate ligands, a variety of structures were obtained. Recently, various compounds with complex topologies based on some ditriazole–pyridine ligands, have been reported by Hou,19 Li,20 Sha,21 and Zhou.22 These ligands revised various conformations, highlighting the fact that they are another class of excellent multi-connection linkers in addition to polycarboxylate ligands.23 Thus, these considerations inspired us to explore new coordination frameworks with this kind of ligand.
In this paper, we introduce a new ditriazole–pyridine ligand: 5,5′-di(pyridin-4-yl)-3,3′-bi(1,2,4-triazole) (4,4′-H2dbpt) which is different to our previous N-heterocyclic-like ligands and introduces a chelating effect between the adjacent triazole rings. Thus, it could adopt various conformations that may lead to unpredictable and interesting structures. By regulating the rotation angles of the four aromatic rings with respect to one another, the deprotonation effort (4,4′-H2dpbt, 4,4′-Hdpbt−, 4,4′-dpbt2−) and the flexing of the angles can be investigated. To the best of our knowledge, MOFs based on 5,5′-di(pyridin-4-yl)-3,3′-bi(1,2,4-triazole) (4,4′-H2dbpt) have not been documented to date.
A series of coordination polymers based on 4,4′-H2dbpt, namely, {[Co5(4,4′-dbpt)2Cl8]·2(C2H5)3NH}n (1), {[Co3(p-BDC)2(4,4′-dbpt)·0.5CH3OH]}n (2), {[Co(4,4′-Hdbpt)2]·H2O}n (3), {[Ni(4,4′-Hdbpt)2]·H2O}n (4), {[Cu2(4,4′-dbpt)]·H2O}n (5), {[Mn(4,4′-dbpt)(H2O)2]·2H2O}n (6) were successfully constructed. By regulating the different auxiliary ligands, central metal ion, 4,4′-H2dbpt displayed five different coordination modes (Scheme 1) based on alteration of the rotation angles, valence state and the flexing angles. As a result, 1–6 adopted five different architectures (3 and 4 are isostructural) with diversiform connectivity from one- to three-dimensional.
 | ||
| Scheme 1 The versatile coordination modes of 4,4′-H2dbpt used in this work. | ||
Experimental section
Materials and physical measurements
With the exception of the ligand 4,4′-H2dbpt that was prepared according to the literature procedure,24 all reagents and solvents for synthesis and analysis were commercially available and used as received. IR spectra were taken on a Perkin-Elmer spectrum One FT-IR spectrometer in the 4000–400 cm−1 region with KBr pellets. Elemental analyses for C, H and N were carried out on a Perkin-Elmer Model 2400 II elemental analyzer. Magnetic susceptibility measurements of the polycrystalline samples were obtained over the temperature range of 2–300 K with a Quantum Design MPMS-XL7 SQUID magnetometer using an applied magnetic field of 1000 Oe. A diamagnetic correction to the observed susceptibilities was applied using Pascal's constants. X-ray powder diffraction (XRPD) intensities were measured on a Rigaku D/max-IIIA diffractometer (Cu-Kα, λ = 1.54056 Å). The crystalline powder samples were prepared by crushing the crystals and spectra scanned from 3 to 60° with a step of 0.1° s−1. Calculated patterns of 1–6 were generated with the PowderCell software.Syntheses of complexes 1–6
X-ray crystallographic determination
All reflection data were collected on an Agilent Supernova diffractometer (Mo, λ = 0.71073 Å) at room temperature. A semi-empirical absorption correction was applied using the SADABS program and the raw data frame integration was performed with SAINT.25 The crystal structures were solved by direct methods using the program SHELXS-97 (ref. 26) and refined by the full-matrix least-squares method on F2 with anisotropic thermal parameters for all non-hydrogen atoms using SHELXL-97.27 All hydrogen atoms were placed in calculated positions and refined isotropically, except the hydrogen atoms of water molecules, which were located in a difference Fourier map and refined isotropically. The details of the crystal data are summarized in Table 1 and selected bond lengths and angles are listed in Table S1 for compounds 1–6 (ESI†).| Complex | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Empirical formula | C40H48Cl8Co5N18 | C30.5H18Co3N8O8.5 | C28H22CoN16O2 | C28H22NiN16O2 | C14H10Cu2N8O | C14H16MnN8O4 |
| Formula weight | 1359.20 | 809.32 | 673.55 | 673.33 | 433.36 | 415.29 |
| Crystal system | Triclinic | Triclinic | Monoclinic | Monoclinic | Orthorhombic | Tetragonal |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/n | P21/n | Pbcn | I41/acd |
| a (Å) | 11.5936 (6) | 9.2529 (12) | 9.2567 (10) | 9.1787 (10) | 14.6978 (14) | 19.1314 (3) |
| b (Å) | 11.9201 (7) | 10.6355 (11) | 15.269 (4) | 15.2181 (11) | 19.3933 (17) | 19.1314 (3) |
| c (Å) | 12.1223 (6) | 11.0493 (13) | 10.2578 (3) | 10.3355 (8) | 10.3560 (9) | 20.6667 (3) |
| α (°) | 63.593 (5) | 63.622 (11) | 90 | 90 | 90 | 90 |
| β (°) | 88.025 (4) | 86.489 (10) | 96.720 (5) | 96.657 (8) | 90 | 90 |
| γ (°) | 67.331 (5) | 74.907 (10) | 90 | 90 | 90 | 90 |
| Volume (Å3) | 1365.40 (12) | 938.68 (19) | 1439.9 (4) | 1433.9 (2) | 2951.9 (5) | 7564.2 (2) |
| Z | 1 | 1 | 2 | 2 | 8 | 16 |
| Calculated density (mg m−3) | 1.651 | 1.432 | 1.554 | 1.559 | 1.941 | 1.459 |
| Goodness-of-fit on F2 | 1.040 | 1.171 | 1.040 | 1.029 | 1.033 | 1.079 |
| Independent reflections | 5578 | 3840 | 2938 | 2922 | 3018 | 1933 |
| Rint | 0.0712 | 0.0249 | 0.0247 | 0.0841 | 0.0521 | 0.0313 |
| R1[I > 2σ(I)] | 0.0652 | 0.0596 | 0.0391 | 0.0530 | 0.0538 | 0.0397 |
| wR2 (all data) | 0.1433 | 0.1933 | 0.0963 | 0.1073 | 0.1390 | 0.1180 |
XRPD results
To confirm whether the crystal structures are truly representative of the bulk materials, X-ray powder diffraction (XRPD) experiments have also been carried out for 1–6. The XRPD experimental and calculated patterns of the corresponding complexes are shown in ESI, Fig. S2.† Although the experimental patterns have a few unindexed diffraction lines and some are slightly broadened in comparison with those calculated from the single crystal models, it can still be considered favorably that the bulk synthesized materials and the crystals as grown are homogeneous for 1–6.Results and discussion
Description of the crystal structures
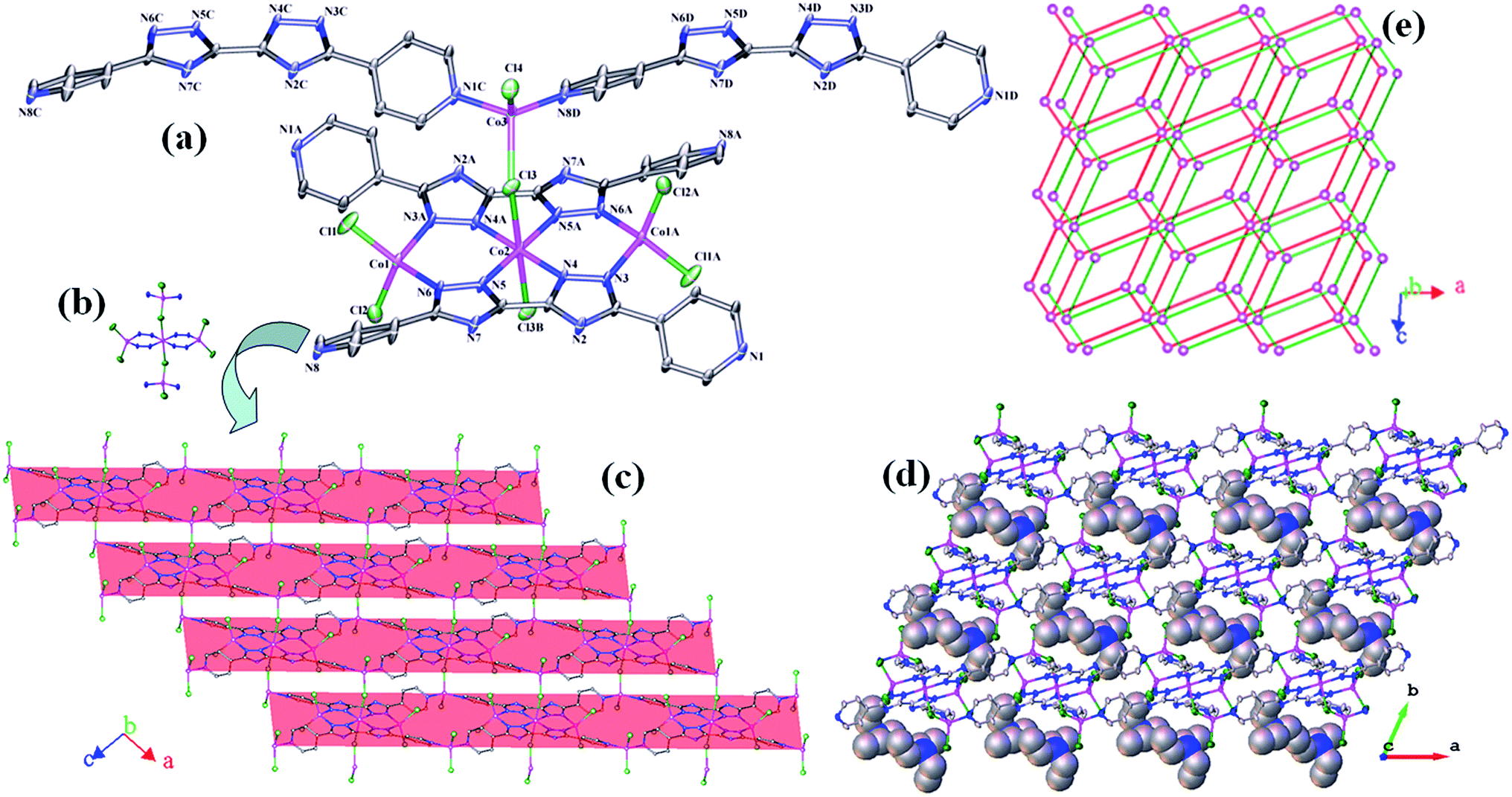 | ||
| Fig. 1 (a) The coordination environments of the Co(II) atoms (symmetry codes: A: x, y, z + 1; B: x + 1, y, z; C: −x + 1, −y + 1, −z + 1; D: x, y, z − 1); (b) the Co5 unit; (c) view of the 2D ladder network; (d) the 3D extended structure; (e) the schematic description for the 2D architecture with (43)3(46.66.83)2 symbol. | ||
 | ||
| Fig. 2 (a) The coordination environments of the Co(II) atoms (symmetry codes: A: −x + 1, −y + 1, −z + 1; B: −x, −y + 1, −z + 1; C: x + 1, y, z; D: −x, −y, −z + 1; E: x − 1, y, z; F: −x + 1, −y, −z + 2); (b) view of the 2D network connected by the 4,4′-dbpt ligands; (c) view of the 3D network along the a axis; (d) the 3D network with parallel channels viewed along the b axis; (e) the schematic description for the 3D architecture with (36.418.53.6) symbol. | ||
 | ||
| Fig. 3 (a) The coordination environment of the M(II) atom (symmetry codes: A: −x + 1, −y, −z + 2; B: x + 1/2, −y − 1/2, z − 1/2; C: −x + 1/2, y + 1/2, −z + 5/2); (b) view of the 2D network connected by the 4,4′-Hdbpt ligand; (c) the 3D extended structure; (d) the schematic description for the 3D architecture with (44.62) symbol. | ||
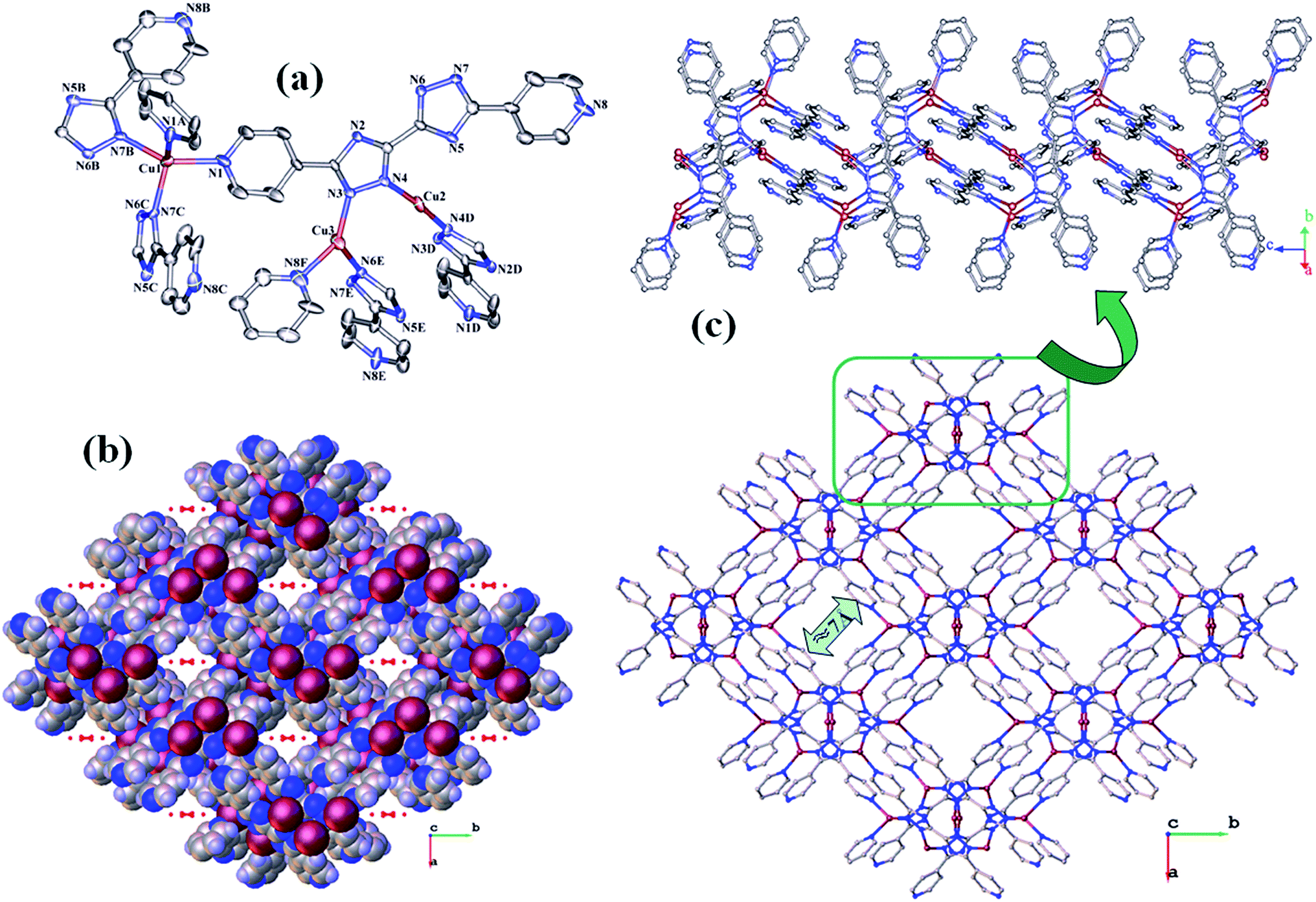 | ||
| Fig. 4 (a) The coordination environments of the Cu(I) atoms (symmetry codes: A: −x, y, −z + 1/2; B: −x + 1/2, y − 1/2, z; C: x − 1/2, y − 1/2, −z + 1/2; D: −x + 1, y, −z + 3/2; E: x, −y + 1, z + 1/2; F: x − 1/2, y − 1/2, −z + 3/2); (b) the space-filling plot of the 3D network connected by the 4,4′-dbpt ligands; (c) view of the 3D network connected by the 4,4′-dbpt ligands. | ||
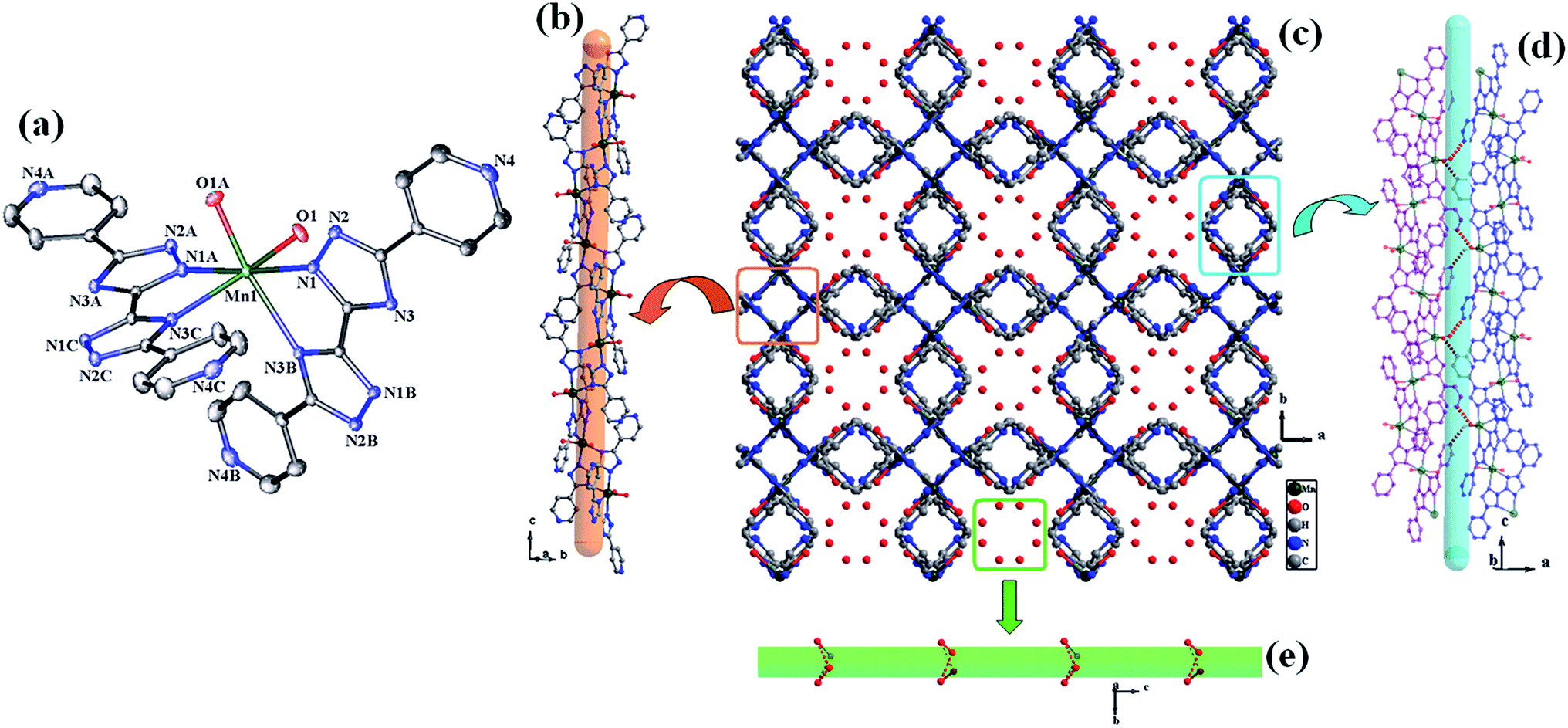 | ||
| Fig. 5 (a) The coordination environment of the Mn(II) atoms (symmetry codes: A: −x + 1/2, y, −z; B: −y + 1/4, −x + 1/4, −z + 1/4; C: y + 1/4, −x + 1/4, z − 1/4); (b) view of the helical chain extended along the c axis; (c) view of the 3D extended structure along the c axis; (d) the connection type of the H-bond between the adjacent helical chains; (e) arrangement of lattice water molecules filling the gaps between neighboring chains. | ||
Structural diversity of 1–6
Six new MOFs, from one to three dimensions, based on the 4,4′-H2dbpt ligand were presented. Among them, 1, 3, and 4 form 2D layer architectures, 2 and 5 form 3D network architectures, and 6 forms a 1D chain architecture with diversiform connectivity. Clearly, the phenomenon of structural diversification in 1–6 may arise from the three sources as follow. First, the 4,4′-H2dbpt ligand plays a dominating effect on constructing the polymer structures. Owing to the different rotation angles of the four aromatic rings with respect to each other, deprotonation effort (4,4′-H2dpbt, 4,4′-Hdpbt−, 4,4′-dpbt2−) and the flexing angles, the ligand could adopt various conformations, which may lead to unpredictable and interesting structures. In this paper, the 4,4′-H2dbpt ligand adopts five different conformations (Scheme 1) according to the geometric requirements of metal ions and/or the introduction of auxiliary ligands. Secondly, the introduction of auxiliary ligands may play a significant role in the formation of different topological structures. Compound 3, with no auxiliary ligand, reveals a 2D, 4-connected topology with a (44.62) Schläfli symbol. For 1, Cl− is involved in coordination and, as a result, a more complex 2D ladder structure based on a Co5 unit with the short Schläfli symbol of (43)3(46.66.83)2 is formed. For 2, with the employment of H2 p-BDC, an 8-connected, 3D pillared layered architecture with the short Schläfli symbol of (36.418.53.6) is formed. Thirdly, different metal ions may have different charges, electron configurations, and ionic radii, and hence exhibit different coordination geometries. Therefore, the connectivity of polymeric frameworks is also strongly related to the metal centers. In 5, Cu(I) exhibits the coexistence of two-, three- and four-coordination patterns, which is different from the others and a complex 3D architecture is formed.Magnetic properties
Magnetic susceptibility measurements were carried out on polycrystalline samples of 1–4 and 6 in the temperature range of 2.0–300.0 K at 1000 Oe (Fig. 6a and 7). | ||
Fig. 6 (a) Plots of χMT vs. T and χM−1 vs. T (inset) for 1; (b) FC magnetization of 1 in different fields; (c) plots of zero-field cooled magnetization (ZFC) and field-cooled magnetization (FCM) and in a field of 10 Oe for 1 using a SQUID; (d) Arrhenius plot for 1 fitted by Arrhenius law τ = τ0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−ΔE/kBT), inset: the zero-field ac magnetic susceptibilities for 1. exp(−ΔE/kBT), inset: the zero-field ac magnetic susceptibilities for 1. | ||
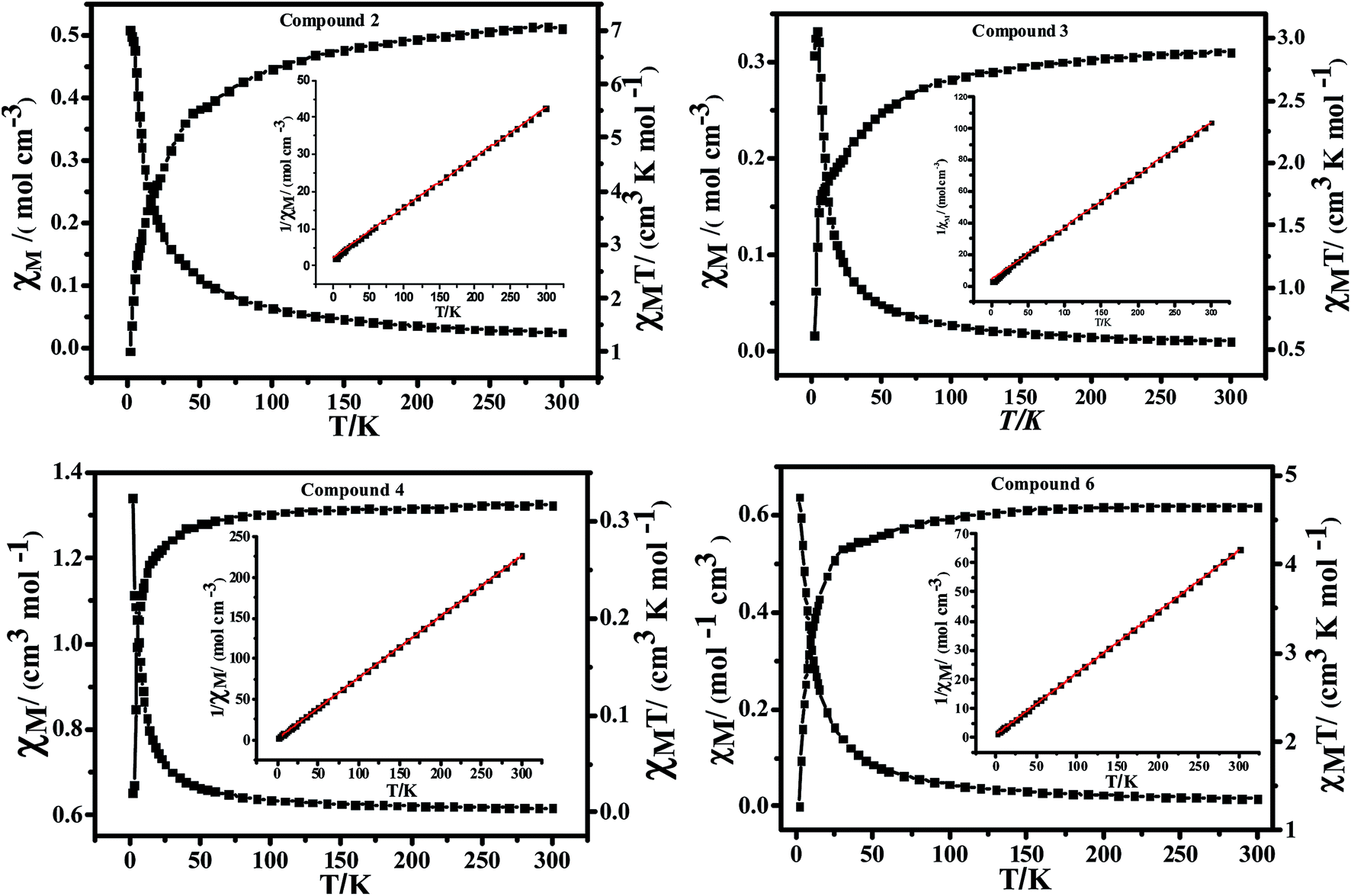 | ||
| Fig. 7 Plots of χMT vs. T and χM−1 vs. T (inset) for 2–4 and 6. | ||
For 1, the data above 50 K follow the Curie–Weiss law with C = 10.81 cm3 K mol−1 and θ = −22.35 K. The χMT value at 300 K is 10.12 cm3 K mol−1, which is larger than the spin-only value 9.38 emu K mol−1 for five magnetically active Co(II) ions (S = 3/2, g = 2.0), as expected for Co(II) systems with a significant contribution from the effects of spin–orbital coupling. As the temperature is lowered, the χMT values decrease continuously and reach a local minimum of 5.98 cm3 K mol−1 at about 12 K, indicative of a strong single-ion behavior admixture with a weak antiferromagnetic interaction,29 and then increase slightly to reach a maximum at ca. 8.0 K, and then rapidly decrease to a minimum of 2.29 cm3 K mol−1 at 2 K. The observed increase in χMT below this temperature is no longer coming from the single-ion behaviour but rather from a ferromagnetic or canted antiferromagnetic Co(II)–Co(II) exchange interaction, and the final decrease in χMT below 10 K indicates a magnetic phase transition. The dependence of χMT vs. T curves of 1 at different fields is pronounced at a low temperature, the larger increase of χMT values being at small fields (Fig. 6b). This is an important feature of spin canting behavior.30
The zero-field ac susceptibility data of 1 shows obvious peaks and slight frequency dependence (Fig. 6d, inset) below 8 K, which confirms the magnetic phase transition. The shift of peak temperature (Tp) of χ′′M for 1 was measured by a parameter ø = ΔTp/[TpΔ(logf)] ≈ 0.06, which is in the range of spin glass systems.31 The relaxation time τ0 was obtained from the Arrhenius law: the best set of parameters is τ0 = 2.53 × 10−18 s and ΔE/kB = 201.55 K (Fig. 6d), where ΔE is the energy barrier and kB is the Boltzmann constant, suggesting a thermally activated mechanism.
For 2, the data above 2 K follow the Curie–Weiss law with C = 7.48 cm3 K mol−1 and θ = −18.35 K. The χMT value at 300 K is 7.05 cm3 K mol−1, which is larger than the spin-only value of 5.63 cm3 K mol−1 for three magnetically active Co(II) ions (S = 3/2, g = 2.0), as expected for Co(II) systems with a significant contribution from the effects of spin–orbital coupling. As the temperature is lowered, the χMT values decrease continuously and reach a local minimum of 1.01 cm3 K mol−1 at about 2 K, indicative of a strong single-ion behavior admixture with a weak antiferromagnetic interaction.
For 3, the data above 2 K follow the Curie–Weiss law with C = 3.01 cm3 K mol−1 and θ = −12.47 K. The χMT value at 300 K is 2.89 cm3 K mol−1, which is much larger than the spin-only value of 1.88 cm3 K mol−1 for a magnetically active Co(II) ion (S = 3/2, g = 2.0), as expected for Co(II) systems with a significant contribution from the effects of spin–orbital coupling. As the temperature is lowered, the χMT values decrease continuously and reach a local minimum of 0.61 cm3 K mol−1 at about 2 K, indicative of a strong single-ion behavior. The distance of the nearest Co atoms in 3 is 9.25 (1) Å, indicating that the weak interactions between them could be ignored. The in- and out-of-phase ac susceptibilities have no dependence on frequency between 2 and 10 K for 3, indicating that there is no single-ion magnet (SIM) behavior in 3 (Fig. S3†).
For 4, the data above 2 K follow the Curie–Weiss law with C = 1.33 cm3 K mol−1 and θ = −1.94 K. The χMT value at 300 K is 1.32 cm3 K mol−1, which is slightly larger than the spin-only value of 1.21 cm3 K mol−1 for a magnetically active Ni(II) ion (S = 1, g = 2.2), and is relatively constant down to approximately 40 K. Below this temperature, it collapses, indicating significant ZFS in the S = 1 ground state.32 Similar to 3, the distance of the nearest Ni atoms in 4 is 9.18 (1) Å, indicating that the weak interactions between them could be ignored. The in- and out-of-phase ac susceptibilities have no dependence on frequency between 2 and 10 K for 4, indicating that there is no SIM behavior in 4 (Fig. S3†).
For 6, the data above 2 K follow the Curie–Weiss law with C = 4.73 cm3 K mol−1 and θ = −4.70 K. The χMT value at 300 K is 4.65 cm3 K mol−1, which is slightly larger than the spin-only value of 4.38 cm3 K mol−1 for a magnetically active Mn(II) ion (S = 5/2, g = 2.2) and is relatively constant down to approximately 30 K. Below this temperature, the χMT values decrease continuously and reach a local minimum of 1.27 cm3 K mol−1 at about 2 K, indicative of a weak antiferromagnetic Mn(II)–Mn(II) exchange interaction. The in- and out-of-phase ac susceptibilities have no dependence on frequency between 2 and 10 K (Fig. S3†).
Thermal analyses
The thermogravimetric (TG) analysis was performed in a N2 atmosphere on polycrystalline samples of complexes 1–6, and the TG curves are shown in Fig. 8. | ||
| Fig. 8 TG curves for complexes 1–6. | ||
For 1, the first weight loss of 15.08% between 30 and 112 °C corresponds to the release of the lattice triethylamine molecules (calculated, 14.89%). The residual framework decomposed beyond 360 °C in a series of complicated weight losses until 920 °C. For 2, the first weight loss of 1.82% between 30 and 85 °C corresponds to the release of the lattice methyl alcohol molecules (calculated, 1.98%). The residual framework decomposed beyond 316 °C in a series of complicated weight losses until 885 °C. For 3, the two steps of the weight loss of 5.14% between 30 and 268 °C correspond to the release of the two lattice water molecules (calculated, 5.34%). The residual framework decomposed beyond 450 °C in a series of complicated weight losses until 844 °C. For 4, similar to 3, the two steps of the weight loss of 5.46% between 30 and 280 °C correspond to the release of the two lattice water molecules (calculated, 5.35%). The residual framework decomposed beyond 428 °C in a series of complicated weight losses until 880 °C. For 5, the first weight loss of 4.16% between 30 and 77 °C corresponds to the release of the lattice water molecules (calculated, 4.13%). The residual framework decomposed beyond 502 °C in a series of complicated weight losses and was still continuing when heating ended at 1000 °C. For 6, the first weight loss of 82.21% between 80 and 184 °C corresponds to the release of the lattice and the coordinated water molecules (calculated, 82.66%). The residual framework decomposed beyond 409 °C in a series of complicated weight losses until 834 °C.
Conclusions
In this paper, we have presented the synthesis and crystal structures of six new MOFs from one to three dimensions based on the 4,4′-H2dbpt ligand. The structural diversities indicate that the 4,4′-H2dbpt ligand could adopt different conformations according to the geometric requirements of metal ions and/or the introduction of auxiliary ligands owing to the different rotation angles of the four aromatic rings with respect to each other, deprotonation effort (4,4′-H2dpbt, 4,4′-Hdpbt−, 4,4′-dpbt2−) and the flexing angles. As a result, five diverse and interesting architectures were obtained. 1 has a 2D (3,6)-topology with (43)3(46.66.83)2 Schläfli symbol. 2 has a 3D 8-connected topology with (36.418.53.6) Schläfli symbol. 3 and 4 which are isostructural, have a 2D 4-connected topology with (44.62) Schläfli symbol. 5 has a complex 3D porous architecture with a 1D solvent-filled channel. 6 reveals a 1D helical chain extended along a 4-fold screw axis. Accordingly, our present findings will further enrich crystal engineering strategy and offer the possibility of controlling the formation of desired network structures.Acknowledgements
We gratefully acknowledge the National Nature Science Foundation of China (nos 21101035, 21361003 and 21461003), the Guangxi Natural Science Foundation of China (2012GXNSFBA053017, 2012GXNSFAA053035, and 2014GXNSFBA118056) and the Foundation of Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources.References
- (a) H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed; (b) Y. Y. Liu, S. Couck, M. Vandichel, M. Grzywa, K. Leus, S. Biswas, D. Volkmer, J. Gascon, F. Kapteijn, J. F. M. Denayer, M. Waroquier, V. V. Speybroeck and P. V. D. Voort, Inorg. Chem., 2013, 52, 113–120 CrossRef CAS PubMed; (c) M. L. Foo, S. Horike, Y. Inubushi and S. Kitagawa, Angew. Chem., Int. Ed., 2012, 51, 6107–6111 CrossRef CAS PubMed; (d) J. H. Luo, H. W. Xu, Y. Liu, Y. S. Zhao, L. L. Daemen, C. Brown, T. V. Timofeeva, S. Q. Ma and H. C. Zhou, J. Am. Chem. Soc., 2008, 130, 9626–9627 CrossRef CAS PubMed; (e) L. Q. Ma, A. Jin, Z. G. Xie and W. B. Lin, Angew. Chem., Int. Ed., 2009, 48, 9905–9908 CrossRef CAS PubMed.
- (a) A. Alberola, E. Coronado, J. R. Galán-Mascarós, C. Giménez-Saiz and C. J. Gómez-García, J. Am. Chem. Soc., 2003, 125, 10774–10775 CrossRef CAS PubMed; (b) H. Okawa, M. Sadakiyo, T. Yamada, M. Maesato, M. Ohba and H. Kitagawa, J. Am. Chem. Soc., 2013, 135, 2256–2262 CrossRef CAS PubMed; (c) U. Subbarao and S. C. Peter, Cryst. Growth Des., 2013, 13, 953–959 CrossRef CAS; (d) E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-Garcia and V. Laukhln, Nature, 2000, 408, 447–449 CrossRef CAS PubMed.
- (a) F. Nouar, J. Eckert, J. F. Eubank, P. Forster and M. Eddaoudi, J. Am. Chem. Soc., 2009, 131, 2864–2870 CrossRef CAS PubMed; (b) X. L. Zhao, X. Y. Wang, S. N. Wang, J. M. Dou, P. P. Cui, Z. Chen, D. Sun, X. P. Wang and D. F. Sun, Cryst. Growth Des., 2012, 12, 2736–2739 CrossRef CAS; (c) J. R. Choi, T. Tachikawa, M. Fujitsuka and T. Majima, Langmuir, 2010, 26, 10437–10443 CrossRef CAS PubMed; (d) C. A. Bauer, T. V. Timofeeva, T. B. Settersten, B. D. Patterson, V. H. Liu, B. A. Simmons and M. D. Allendorf, J. Am. Chem. Soc., 2007, 129, 7136–7144 CrossRef CAS PubMed; (e) K. A. White, D. A. Chengelis, K. A. Gogick, J. Stehman, N. L. Rosi and S. Petoud, J. Am. Chem. Soc., 2009, 131, 18069–18071 CrossRef CAS PubMed; (f) J. R. Li, Y. Tao, Q. Yu and X. H. Bu, Chem. Commun., 2007, 1527–1529 RSC; (g) B. Dey, R. Saha and P. Mukherjee, Chem. Commun., 2013, 49, 7064–7066 RSC.
- (a) Z. M. Wang, Y. J. Zhang, T. Liu, M. Kurmoo and S. Gao, Adv. Funct. Mater., 2007, 17, 1523–1536 CrossRef CAS PubMed; (b) Z. R. Pan, H. G. Zheng, T. W. Wang, Y. Song, Y. Z. Li, Z. J. Guo and S. R. Batten, Inorg. Chem., 2008, 47, 9528–9536 CrossRef CAS PubMed; (c) J. P. Zhao, Q. Yang, Z. Y. Liu, R. Zhao, B. W. Hu, M. Du, Z. Chang and X. H. Bu, Chem. Commun., 2012, 48, 6568–6570 RSC; (d) N. Yanai, W. Kaneko, K. Yoneda, M. Ohba and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 3496–3497 CrossRef CAS PubMed; (e) P. Lama, J. Mrozinski and P. K. Bharadwaj, Cryst. Growth Des., 2012, 12, 3158–3168 CrossRef CAS; (f) S. H. Cho, B. Ma, S. T. Nguyen, J. T. Hupp and T. E. Albrecht-Schmitt, Chem. Commun., 2006, 2563–2565 RSC; (g) D. Pinkowicz, Z. Y Li, P. Pietrzyk and M. Rams, Cryst. Growth Des., 2014, 14, 4878–4881 CrossRef CAS.
- (a) M. Y. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed; (b) D. W. Feng, Z. Y. Gu, J. R. Li, H. L. Jiang, Z. W. Wei and H. C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed; (c) J. M. Roberts, B. M. Fini, A. A. Sarjeant, O. K. Farha, J. T. Hupp and K. A. Scheidt, J. Am. Chem. Soc., 2012, 134, 3334–3337 CrossRef CAS PubMed; (d) Z. L. Liao, G. D. Li, M. H. Bi and J. S. Chen, Inorg. Chem., 2008, 47, 4844–4853 CrossRef CAS PubMed; (e) J. M. Zhang, A. V. Biradar, S. Pramanik, T. J. Emge, T. Asefa and J. Li, Chem. Commun., 2012, 48, 6541–6543 RSC; (f) L. Q. Ma, C. Abney and W. B. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- (a) O. D. Friedrichs, M. O'Keeffe and O. M. Yaghi, Acta Crystallogr., Sect. A: Found. Crystallogr., 2003, 59, 22 CrossRef PubMed; (b) O. D. Friedrichs, M. O'Keeffe and O. M. Yaghi, Acta Crystallogr., Sect. A: Found. Crystallogr., 2003, 59, 515 CrossRef PubMed; (c) L. Pan, D. H. Olson, L. R. Ciemnolonski, R. Heddy and J. Li, Angew. Chem., Int. Ed., 2006, 45, 616–619 CrossRef CAS PubMed; (d) A. Y. Robin and K. M. Fromm, Coord. Chem. Rev., 2006, 250, 2127–2157 CrossRef CAS PubMed; (e) C. Pan, J. P. Nan, X. L. Dong, X. M. Ren and W. Q. Jin, J. Am. Chem. Soc., 2011, 133, 12330–12333 CrossRef CAS PubMed; (f) X. Z. Song, S. Y. Song, C. Qin, S. Q. Su, S. N. Zhao, M. Zhu, Z. M. Hao and H. J. Zhang, Cryst. Growth Des., 2012, 12, 253–263 CrossRef CAS.
- (a) Y.-L. Wang, D.-Q. Yuan, W.-H. Bi, X. Li, X.-J. Li, F. Li and R. Cao, Cryst. Growth Des., 2005, 5, 1849–1855 CrossRef CAS; (b) Y.-B. Lu, M.-S. Wang, W.-W. Zhou, G. Xu, G.-C. Guo and J.-S. Huang, Inorg. Chem., 2008, 47, 8935–8942 CrossRef CAS PubMed; (c) O. Fabelo, J. Pasan, F. Lloret, M. Julve and C. R. Perez, Inorg. Chem., 2008, 47, 3568–3576 CrossRef CAS PubMed.
- (a) W.-G. Lu, L. Jiang, X.-L. Feng and T.-B. Lu, Cryst. Growth Des., 2008, 8, 986–994 CrossRef CAS; (b) A.-X. Tian, J. Ying, J. Peng, J.-Q. Sha, H.-J. Pang, P.-P. Zhang, Y. Chen, M. Zhu and Z.-M. Su, Inorg. Chem., 2009, 48, 100–110 CrossRef CAS PubMed.
- (a) Y. Wang, X.-Q. Zhao, W. Shi, P. Cheng, D.-Z. Liao and S.-P. Yan, Cryst. Growth Des., 2009, 9, 2137–2145 CrossRef CAS; (b) J. Park, S. Hong, D. Moon, M. Park, K. Lee, S. Kang, Y. Zou, R. P. John, G. H. Kim and M. S. Lah, Inorg. Chem., 2007, 46, 10208–10213 CrossRef CAS PubMed; (c) C.-H. Li, K.-L. Huang, Y.-N. Chi, X. Liu, Z.-G. Han, L. Shen and C.-W. Hu, Inorg. Chem., 2009, 48, 2010–2017 CrossRef CAS PubMed.
- (a) L. S. Long, CrystEngComm, 2010, 12, 1354–1365 RSC; (b) M. Tsaramyrsi, M. Kaliva, A. Salifoglou, C. P. Raptopoulou, A. Terzis, V. Tangoulis and J. Giapintzakis, Inorg. Chem., 2001, 40, 5772–5779 CrossRef CAS PubMed; (c) S. Faulkner and B. P. Burton-Pye, Chem. Commun., 2005, 259–261 RSC; (d) L. N. Zhang, S. T. Lu, C. Zhang, C. X. Du and H. W. Hou, CrystEngComm, 2015, 17, 846–855 RSC.
- (a) Y. Tao, J.-R. Li, Q. Yu, W.-C. Song, X.-L. Tong and X.-H. Bu, CrystEngComm, 2008, 10, 699–705 RSC; (b) B. Zheng, H. Dong, J. Bai, Y. Li, S. Li and M. Scheer, J. Am. Chem. Soc., 2008, 130, 7778–7779 CrossRef CAS PubMed.
- (a) H. Furukawa, Y. B. Go, N. Ko, Y. K. Park, F. J. Uribe- Romo, J. Kim, M. O'Keeffe and O. M. Yaghi, Inorg. Chem., 2011, 50, 9147–9152 CrossRef CAS PubMed; (b) J. J. Gassensmith, H. Furukawa, R. A. Smaldone, R. S. Forgan, Y. Y. Botros, O. M. Yaghi and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 15312–15315 CrossRef CAS PubMed; (c) K. M. Choi, H. J. Jeon, J. K. Kang and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11920–11923 CrossRef CAS PubMed; (d) A. Phan, A. U. Czaja, F. Gándara, C. B. Knobler and O. M. Yaghi, Inorg. Chem., 2011, 50, 7388–7390 CrossRef CAS PubMed; (e) J. Song, Z. Luo, D. K. Britt, H. Furukawa, O. M. Yaghi, K. I. Hardcastle and C. L. Hill, J. Am. Chem. Soc., 2011, 133, 16839–16846 CrossRef CAS PubMed.
- (a) R. B. Getman, J. H. Miller, K. Wang and R. Q. Snurr, J. Phys. Chem. C, 2011, 115, 2066–2075 CrossRef CAS; (b) X. Y. Bao, R. Q. Snurr and L. J. Broadbelt, Langmuir, 2009, 25, 10730–10736 CrossRef CAS PubMed.
- (a) X. M. Chen and G. F. Liu, Chem.–Eur. J., 2002, 8, 4811–4817 CrossRef CAS; (b) J. P. Zhang, Y. Y. Lin, X. C. Huang and X. M. Chen, J. Am. Chem. Soc., 2005, 127, 5495–5506 CrossRef CAS PubMed; (c) L. Cheng, W. X. Zhang, B. H. Ye, J. B. Lin and X. M. Chen, Inorg. Chem., 2007, 46, 1135–1143 CrossRef CAS PubMed; (d) J. P. Zhang, X. L. Qi, Z. J. Liu, A. X. Zhu, Y. Chen, J. Wang and X. M. Chen, Cryst. Growth Des., 2011, 11, 796–802 CrossRef CAS; (e) J. B. Lin, J. P. Zhang, W. X. Zhang, W. Xue, D. X. Xue and X. M. Chen, Inorg. Chem., 2009, 48, 6652–6660 CrossRef CAS PubMed.
- (a) C. P. Li, J. M. Wu and M. Du, Inorg. Chem., 2011, 50, 9284–9289 CrossRef CAS PubMed; (b) L.-F. Ma, C.-P. Li, L.-Y. Wang and M. Du, Cryst. Growth Des., 2010, 10, 2641–2649 CrossRef CAS; (c) C.-P. Li, Q. Yu, J. Chen and M. Du, Cryst. Growth Des., 2010, 10, 2650–2660 CrossRef CAS; (d) M. Du, Q. Wang, C.-P. Li, X.-J. Zhao and J. Ribas, Cryst. Growth Des., 2010, 10, 3285–3289 CrossRef CAS.
- (a) Z. Yan, Z. P. Ni, F. S. Guo, J. Y. Li, Y. C. Chen, J. L. Liu, W. Q. Lin, D. Aravena, E. Ruiz and M. L. Tong, Inorg. Chem., 2014, 53, 201–208 CrossRef CAS PubMed; (b) Z. Yan, J. Y. Li, T. Liu, Z. P. Ni, Y. C. Chen, F. S. Guo and M. L. Tong, Inorg. Chem., 2014, 53, 8129–8135 CrossRef CAS PubMed.
- (a) X. T. Zhang, L. M. Fan, Z. Sun, W. Zhang, D. C. Li, J. M. Dou and L. Han, Cryst. Growth Des., 2013, 13, 792–803 CrossRef CAS; (b) L. M. Fan, X. T. Zhang, Z. Sun, W. Zhang, Y. S. Ding, W. L. Fan, L. M. Sun, X. Zhao and H. Lei, Cryst. Growth Des., 2013, 13, 2462–2475 CrossRef CAS.
- (a) F.-P. Huang, J.-L. Tian, W. Gu, X. Liu, S.-P. Yan, D.-Z. Liao and P. Cheng, Cryst. Growth Des., 2010, 10, 1145–1154 CrossRef CAS; (b) F.-P. Huang, H.-Y. Li, Q. Yu, H.-D. Bian, J.-L. Tian, S.-P. Yan, D.-Z. Liao and P. Cheng, CrystEngComm, 2012, 14, 4756–4766 RSC; (c) F.-P. Huang, J.-L. Tian, D.-D. Li, G.-J. Chen, W. Gu, S.-P. Yan, X. Liu, D.-Z. Liao and P. Cheng, Inorg. Chem., 2010, 49, 2525–2529 CrossRef CAS PubMed; (d) F.-P. Huang, Q. Zhang, Q. Yu, H.-D. Bian, H. Liang, S.-P. Yan, D.-Z. Liao and P. Cheng, Cryst. Growth Des., 2012, 12, 1890–1898 CrossRef CAS; (e) F. P. Huang, P. F. Yao, Q. Yu, D. Yao, H. D. Bian, H. Y. Li, J. L. Tian and S. P. Yan, Inorg. Chem. Commun., 2013, 31, 18–22 CrossRef CAS PubMed; (f) F. P. Huang, Z. M. Yang, P. F. Yao, Q. Yu, J. L. Tian, H. D. Bian, S. P. Yan, D. Z. Liao and P. Cheng, CrystEngComm, 2013, 15, 2657–2668 RSC; (g) F. P. Huang, P. F. Yao, W. Luo, H. Y. Li, Q. Yu, H. D. Bian and S. P. Yan, RSC Adv., 2014, 4, 43641–43652 RSC; (h) F.-P. Huang, J.-L. Tian, D.-D. Li, G.-J. Chen, W. Gu, S.-P. Yan, X. Liu, D.-Z. Liao and P. Cheng, CrystEngComm, 2010, 12, 395–400 RSC.
- (a) Z. Q. Xu, Q. Wang, H. J. Li, W. Meng, Y. Han, H. W. Hou and Y. T. Fan, Chem. Commun., 2012, 48, 5736–5738 RSC; (b) Z. Q. Xu, W. Meng, H. J. Li, H. W. Hou and Y. T. Fan, Inorg. Chem., 2014, 53, 3260–3262 CrossRef CAS PubMed; (c) Z. Q. Xu, H. J. Li, A. R. Li, W. Meng, H. W. Hou and Y. T. Fan, Inorg. Chem. Commun., 2013, 36, 126–129 CrossRef CAS PubMed.
- (a) W. W. Dong, D. S. Li, J. Zhao, L. F. Ma, Y. P. Wu and Y. P. Duan, CrystEngComm, 2013, 15, 5412–5416 RSC; (b) W. W. Dong, D. S. Li, J. Zhao, Y. P. Wu and Y. Y. Wang, Inorg. Chem. Commun., 2012, 21, 53–56 CrossRef CAS PubMed.
- M. T. Li, J. Q. Sha, X. M. Zong, J. W. Sun, P. F. Yan, L. Li and X. N. Yang, Cryst. Growth Des., 2014, 14, 2794–2802 CAS.
- Y. H. Zhou, W. Q. Wan, D. L. Sun, J. Tao, L. Zhang and X. W. Wei, Z. Anorg. Allg. Chem., 2014, 640(1), 249–253 CrossRef CAS PubMed.
- (a) J. P. Zhang, Y. B. Zhang, J. B. Lin and X. M. Chen, Chem. Rev., 2012, 112, 1001 CrossRef CAS PubMed; (b) J. G. Haasnoot, Coord. Chem. Rev., 2000, 200–202, 131–185 CrossRef CAS; (c) T.-F. Liu, J. Lü, C. B. Tian, M. N. Cao, Z. J. Lin and R. Cao, Inorg. Chem., 2011, 50, 2264–2271 CrossRef CAS PubMed.
- N. N. Vyatsheslav, V. Z. Nikolay and Z. V. Sergey, ARKIVOC, 2005, 118 Search PubMed.
- ]SAINT Software Reference Manual, Bruker AXS, Madison, WI, 1998 Search PubMed.
- G. M. Sheldrick, Phase Annealing in SHELX-90: Direct Methods for Larger Structures, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, SHELXS-97, Program for X-ray Crystal Structure Solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- (a) M. O'Keeffe and O. M. Yaghi, Reticular Chemistry Structure Resource, Arizona State University, Tempe, AZ, 2005, http://www.%20okeeffews1.%201a.asu.edu/rcsr/home.htm Search PubMed; (b) V. A. Blatov, Multipurpose crystallochemical analysis with the program package TOPOS, IUCr CompComm Newsletter, 2006, vol. 7, p. 4 Search PubMed; (c) V. A. Blatov, A. P. Shevchenko and V. N. Serezhkin, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS; (d) A. F. Wells, Three-Dimensional Nets and Polyhedra, Wiley Interscience, New York, 1977 Search PubMed.
- (a) L.-H. Jia, R.-Y. Li, Z.-M. Duan, S.-D. Jiang, B.-W. Wang, Z.-M. Wang and S. Gao, Inorg. Chem., 2011, 50, 144–254 CrossRef CAS PubMed; (b) N. Marino, O. F. Ikotun, M. Julve, F. Lloret, J. Cano and R. P. Doyle, Inorg. Chem., 2011, 50, 378–389 CrossRef CAS PubMed; (c) T. D. Keene, I. Zimmermann, A. Neels, O. Sereda, J. Hauser, M. Bonin, M. B. Hursthouse, D. J. Price and S. Decurtins, Dalton Trans., 2010, 4937–4950 RSC; (d) L.-Q. Wei, B.-W. Li, S. Hu and M.-H. Zeng, CrystEngComm, 2011, 13, 510–516 RSC; (e) S.-Q. Zang, X.-M. Ren, Y. Su, Y. Song, W.-J. Tong, Z.-P. Ni, H.-H. Zhao, S. Gao and Q.-J. Meng, Inorg. Chem., 2009, 48, 9623–9630 CrossRef CAS PubMed.
- (a) X.-Y. Wang, H.-Y. Wei, Z.-M. Wang, Z.-D. Chen and S. Gao, Inorg. Chem., 2005, 44, 572–583 CrossRef CAS PubMed; (b) E. K. Brechin, O. Cador, A. Caneschi, C. Cadiou, S. G. Harris, S. Parsons, M. Vonci and R. E. P. Winpenny, Chem. Commun., 2002, 1860–1861 RSC; (c) A. Escuer, J. Cano, M. A. S. Goher, Y. Journaux, F. Lloret, F. A. Mautner and R. Vicente, Inorg. Chem., 2000, 39, 4688–4695 CrossRef CAS; (d) X.-N. Cheng, W. Xue, W.-X. Zhang and X.-M. Chen, Chem. Mater., 2008, 20, 5345–5350 CrossRef CAS; (e) F.-P. Huang, J.-L. Tian, D.-D. Li, G.-J. Chen, W. Gu, S.-P. Yan, X. Liu, D.-Z. Liao and P. Cheng, Inorg. Chem., 2010, 49, 2525–2529 CrossRef CAS PubMed.
- (a) J. A. Mydosh, Spin Glasses: An Experimental Introduction, Taylor & Francis, London, 1993 Search PubMed; (b) X.-N. Cheng, W. Xue, J.-B. Lina and X.-M. Chen, Chem. Commun., 2010, 246–248 RSC.
- R. Ruamps, R. Maurice, L. Batchelor, M. Boggio-Pasqua, R. Guillot, A. L. Barra, J. J. Liu, E. E. Bendeif, S. Pillet, S. Hill, T. Mallah and N. Guihéry, J. Am. Chem. Soc., 2013, 135, 3017–3026 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1033515–1033520. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra06273j |
| This journal is © The Royal Society of Chemistry 2015 |