
Design, synthesis and in vivo antitumor efficacy of novel eight-arm-polyethylene glycol–pterostilbene prodrugs
Abstract
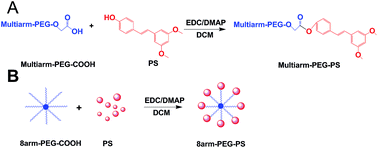
Pterostilbene (PS) is an effective antitumor drug, but its clinical development has been hampered due to its poor solubility and relatively short half-life. To deal with this predicament, an eight-arm-polyethylene glycol–PS (8arm-PEG–PS) prodrug was synthesized by linking PS with eight-arm-polyethylene glycol (8arm-PEG). The obtained 8arm-PEG–PS prodrug possesses high solubility (∼147 fold of free PS) and relatively high drug-binding capacity (∼7.06 wt%). An in vitro cytotoxicity test demonstrated the excellent anticancer activities with a potency similar to that of free PS. In addition, the PS prodrugs significantly improved the therapeutic effect on the inhibition of tumor growth with lower systemic toxicity compared to the native PS. Compared to the group treated by the free PS, the tumor volume of the groups treated by the PS prodrugs decreased dramatically by more than 2 fold or 3 fold in the Lewis lung carcinoma (LLC) tumor-bearing mouse model. On the basis of these results, the 8arm-PEG–PS prodrugs have great promise toward cancer therapy.
 Please wait while we load your content...
Please wait while we load your content...

