
Metal-free activation of peroxymonosulfate by g-C3N4 under visible light irradiation for the degradation of organic dyes
Yufang Taoa,
Qian Nib,
Mingyu Weia,
Dongsheng Xiaa,
Xiaoxia Li*b and
Aihua Xu*a
aSchool of Environmental Engineering, Wuhan Textile University, Wuhan 430073, China. E-mail: lixxwh@163.com; xahspinel@sina.com; Fax: +86-27-59367334; Tel: +86-27-59367334
bSchool of Chemistry and Chemical Engineering, Wuhan Textile University, Wuhan 430073, China
First published on 11th May 2015
Abstract
Semiconducting carbon nitride materials (g-C3N4) were investigated as metal-free catalysts for the activation of peroxymonosulfate (PMS) under visible light irradiation to degrade organic dyes in aqueous solution. X-ray diffraction, scanning electron microscopy, N2 adsorption/desorption isotherms, Fourier transform infrared spectroscopy and X-ray photoelectron spectroscopy were used to characterize the properties of the material. The effect of several parameters including the concentration of catalyst, PMS and organic dye, and initial solution pH on its catalytic activity was also investigated. It was found that the obtained g-C3N4 can effectively activate PMS under visible light irradiation to generate strong sulfate radicals which was highly active for Acid Orange 7 (AO7) and other organic dyes degradation. The catalyst also presented a long-term stability during multiple runs. Based on intermediate detections, the degradation pathway of AO7 in the g-C3N4/PMS/Vis system was proposed. This study demonstrated a promising approach for the activation of green oxidant, PMS, by the newly-developed polymer photocatalysts for environmental remediation and oxidation catalysis.
1. Introduction
Advanced oxidation processes (AOPs) are used as an effective treatment option for the degradation of emerging recalcitrant organic contaminants in water, wastewater treatment, and other environmental applications.1 AOPs can effectively mineralize organic matter into carbon dioxide, water and other inorganic ionic such as chloride and nitrate via the generation of reactive oxygen species.2 Typical AOPs include photodegradation, catalytic ozonation, ultraviolet irradiation, and Fenton.3–6 Sulfate radicals based advanced oxidation technologies has recently received much attention for its capability of destructing many organic pollutants, such as cyanobacterial toxin, microcystin-LR.7 The radical can be generated by activation of persulfate or peroxymonosulfate (PMS) by heat,7–9 UV and transition metal ions10–12 with the oxidation–reduction potential of +2.5–+3.1 V. However, UV activation and thermal activation both need added energy. Transition metal ions catalysts such as Co(II), Cu(II), Mn(II), Ni(II) and Ce(III) are toxic and difficult to recycle. In recent years, the heterogeneous activation of PMS to degrade contaminants has become a more focused area. For example, CuFe2O4 magnetic nanoparticles have been suggested as efficient heterogeneous catalysts for PMS activation.13 In our previous works, it was also found that cryptomelane-type manganese oxide octahedral molecular sieves showed excellent activity and stability for organic dyes decompositions in aqueous solution in the presence of PMS.14,15More recently, activation of PMS to degrade contaminants by metal-free catalysts has attracted the attention of more people. For example, Huang et al. found that the degradation of a typical organic contaminant p-CBA, was significantly enhanced by the addition of O3/PMS.16 In the study of Yang's group, granular activated carbon (GAC) was used as a catalyst to activate peroxymonosulfate (PMS) to degrade azo dye Acid Orange 7 (AO7) in aqueous solution.17 Lee et al. found that carbon nanotubes (CNTs) could activate persulfates (i.e. peroxymonosulfate and peroxydisulfate) into reactive species that are capable of oxidizing organic compounds in water.18 Compared with metal-based catalysts, the metal-free catalyst has advantages of good chemical stability, no heavy metal pollution and environmental friendliness.
Graphitic carbon nitride (g-C3N4) is an Earth-abundant and low-cost photocatalyst capable of degrading organic pollutions or generating H2 and H2O2 from water even in the absence of catalytic metals.19 It is commercially available and can be easily fabricated. Many works based on organic dye degradation, like rhodamine B (RhB),20 methyl orange,21,22 methylene blue,23 and hydrogen evolution,24 have been devoted to study the photocatalytic performance of g-C3N4 and modified g-C3N4. In addition, the addition of oxidants can increases the oxidation rate of organic compounds and avoids problems caused by low oxygen concentration. For example, the g-C3N4 catalyst was also reported to activate hydrogen peroxide under visible light irradiation for the degradation of RhB.25 However, until now there are still no investigations on activation of PMS by g-C3N4 under visible light irradiation for the degradation of organic pollutants. In this article, we report on the advancement of g-C3N4 photocatalysts for environmental application in the presence of PMS. This heterogeneous catalysis method seems to be an economically attractive and environmentally friendly oxidation technology for the treatment of organic pollutants. The reaction mechanism and the effect of several major facts were also discussed.
2. Materials and methods
2.1. Preparation of g-C3N4 catalyst
The photocatalyst of g-C3N4 was prepared by a method of thermal decomposition of melamine similar to that reported on the literature.26 The reaction of heating melamine was carried out in the semiclosed system to prevent sublimation of melamine. 20 g of melamine powder was put into an alumina crucible with a cover, then heated to 500 °C in a muffle furnace for 1 h at a heating rate of 3 °C min−1; then quickly heated to 520 °C in five minutes and maintain this temperature for one hour. The sample was then cooled to room temperature before removing out from the furnace. Yellow products were obtained after grinding and screening.2.2. Characterization
X-ray powder diffraction (XRD) pattern was obtained on a Bruker D8 powder X-ray Diffractometer with Cu Kα radiation (λ = 0.15406 nm). The beam voltage and current used were 40 kV and 40 mA, respectively. Fourier transform infrared (FT-IR) spectra were recorded on a Bruker Vector 22 spectrometer. The sample was mixed with solid KBr, then ground into powder and dried before pressed into a KBr wafer. The surface morphology was characterized on a Hitachi S-4800 scanning electron microscope (SEM) instrument (Hitachi Ltd., Japan) and field-emission transmission electron microscope (FETEM, Tecnai G2 F20). UV-vis diffuse reflectance spectra were recorded on a Shimadzu UV-2450 spectrometer in the range of 200–800 nm with BaSO4 as a reference.The determination of pore size distribution, BET surface area, and pore volume was carried out with a Quanta chrome Autosorb1 at −196 °C. The samples were first degassed at 200 °C for 6 h. The surface area of the samples was obtained using the N2 adsorption isotherm with the Brunauer–Emmett–Teller equation. The average pore diameter was calculated with the Barrett–Joyner–Halenda desorption isotherm. The chemical species in g-C3N4 were investigated by X-ray photoelectron spectra (XPS) on a VG Multilab2000 spectrometer (Thermo Electron Corporation) with Al Kα radiation as the exciting source (300 W). Charging effects were corrected by adjusting the binding energy of C 1s to 284.6 eV.
2.3. Photocatalytic degradation experiment
Degradation experiments were performed in a 100 mL reactor at about 25 °C without irradiation or under visible light irradiation by a 500 W xenon lamp with a stable optical energy flux of about 50 mW cm −2 (Beijing Trusttech Co.). After the desired amounts of AO7 and PMS (Oxone®, DuPont's triple salt: 2KHSO5·KHSO4·K2SO4, Aladdin) in 50 mL of the aqueous solution were added into the reactor, the reaction was initialized by adding g-C3N4. Each reaction solution was constantly stirred with a PTFE coated magnetic stirrer. Since PMS is an acidic oxidant, the addition of PMS led to a significant decrease of pH, and the experiment was conducted at acidic medium (pH 3.38, no adjustment). For studying the effect of solution pH on the rate of AO7 degradation, H2SO4 (20 mM) and NaOH (20 mM) was used to adjust the solution pH after PMS was added into the solution. For the recycling experiment, the catalyst was separated without any treatment after each recycle, and then the next reaction was started by adding a fresh solution of AO7 and PMS.To monitor the degradation process of AO7, solution samples were taken out at given time intervals and measured immediately on a Varian Cary 50 Scan UV-vis spectrophotometer under the maximum absorption wavelength (484 nm). For identification of degradation products, the samples were analyzed by mass spectrometry, and the experiments were performed on an Esquire LC-ion trap mass spectrometer (Bruker Daltonics, Bremen, Germany) equipped with an orthogonal geometry ESI source. Nitrogen was used as the drying (3 L min−1) and nebulizing (6 psi) gas at 300 °C. The spray shield was set to 4.0 kV and the capillary cap was set to 4.5 kV. Scanning was performed from m/z 70 to 800 in the standard resolution mode at a scan rate of 13 kDa s−1. Before analysis, each sample was diluted ten times.
3. Results and discussion
3.1. Characterization of g-C3N4
X-ray diffraction patterns of the g-C3N4 catalysts are shown in Fig. 1(A). The intense peak focused on 27.4° is the typical graphite interlayer, which is marked as 002 peak and the interplanar distance was d = 0.326 nm.27 Fig. 1(B) displays the FT-IR spectra of g-C3N4. The broad absorption band at 3100–3300 cm−1 can be assigned to the stretching modes of secondary and primary amines and their intermolecular hydrogen-bonding interactions. The band at 810 cm−1 belongs to s-triazine ring modes and bands at 1200–1600 cm−1 are characteristic of aromatic carbon nitride heterocycles.28 | ||
| Fig. 1 XRD (A) and FT-IR (B) spectra of fresh and used g-C3N4 catalysts. | ||
The morphological features of g-C3N4 were studied using SEM and TEM. As observed in Fig. 2, the catalyst shows typical flat layer structure with the size of about several micrometers, similar with that reported in literature.29
 | ||
| Fig. 2 SEM (1) and TEM (2) images of fresh g-C3N4 catalyst. | ||
To examine the surface area, as well as the pore size distribution, nitrogen adsorption/desorption analysis was conducted on g-C3N4. The surface area was found to be 13.7 m2 g−1, which was much smaller than that of common photocatalysts or catalyst supports. The isotherms and Barrett–Joyner–Halenda pore size distributions are shown in Fig. 3(A). The isotherms show typical hysteresis, revealing the existence of mesopores, and the pore size mainly distributed in the range of 3–60 nm. The UV-vis diffuse reflectance spectrum of the g-C3N4 powder is shown in Fig. 3(B). It is observed that the g-C3N4 powder exhibits remarkable absorbance in UV region and also strong absorption of visible light within 475 nm, due to the visible-light induced transition of electrons from the valence band to the conduction band of the carbon nitride semiconductor, consistent with the reported results.19
 | ||
| Fig. 3 (A) N2 adsorption/desorption isotherm curve of fresh g-C3N4 catalyst (inset is the pore size distribution of g-C3N4) and (B) UV-vis diffuse reflectance spectrum of fresh g-C3N4 catalyst. | ||
The composition of the obtained g-C3N4 was evaluated by XPS measurements. The C1s XPS spectrum in Fig. 4(A) shows two C1s peaks centering at 284.6 eV and 288.0 eV, corresponding to graphitic carbon and sp2 bonded carbon (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), respectively.30 As shown in Fig. 4(B), the high resolution XPS spectrum of N1s can be deconvoluted into three different components. The peaks at 398.8 eV and 400 eV can be attributed to the sp2 hybridized aromatic nitrogen atoms involved in triazine rings (CN–C) and bridging nitrogen atoms N–(C)3, respectively.30 Another weak peak at 401 eV was also observed, indicating amino functions are carrying hydrogen (C–N–H).30 In addition, the peak at 404.8 eV is assigned to the charging effects or positive charge localization in heterocycles.31
N), respectively.30 As shown in Fig. 4(B), the high resolution XPS spectrum of N1s can be deconvoluted into three different components. The peaks at 398.8 eV and 400 eV can be attributed to the sp2 hybridized aromatic nitrogen atoms involved in triazine rings (CN–C) and bridging nitrogen atoms N–(C)3, respectively.30 Another weak peak at 401 eV was also observed, indicating amino functions are carrying hydrogen (C–N–H).30 In addition, the peak at 404.8 eV is assigned to the charging effects or positive charge localization in heterocycles.31
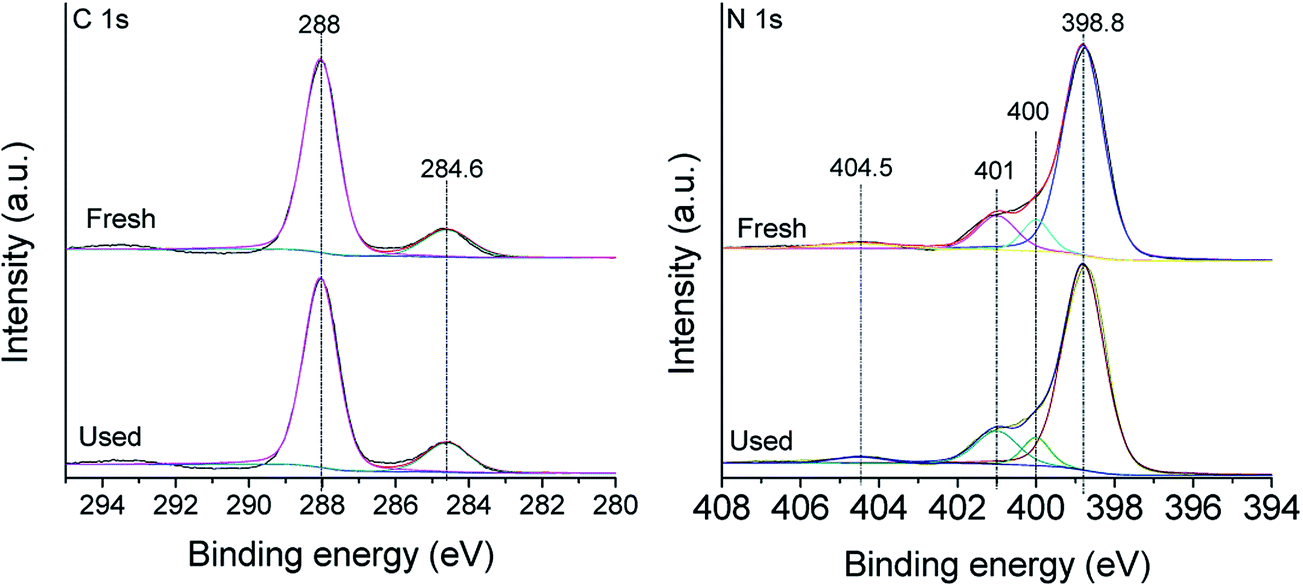 | ||
| Fig. 4 High-resolution XPS spectra of C1s and N1s of fresh and used g-C3N4. | ||
3.2. Photocatalytic performance of g-C3N4
The photocatalytic performance of g-C3N4 for AO7 degradation in different systems was given in Fig. 4(A). Control experiments showed that the removal of AO7 in the PMS or PMS/Vis systems without the catalyst was negligible, indicating that PMS could not degrade the azo dye directly and not be activated by visible light irradiation. For the experiment with g-C3N4 under visible light irradiation, a slight enhancement of AO7 removal was observed, in good agreement with that the catalyst can be used as photocatalyst for organic compound degradation.19 For the g-C3N4/PMS/Vis system, a faster degradation could be obtained and about 86% of the dye was removed during the reaction, while g-C3N4 can not active PMS in the dark, as in the g-C3N4/PMS system the removal of AO7 was only 5%. The result suggested that g-C3N4 could activate PMS under visible light irradiation to produce reactive species such as SO4−˙ to induce strong AO7 degradation. In addition, the activity of the g-C3N4/PMS/Vis system for degradations of other organic dyes with different chemical structures such as the anthraquinone dye KN-R, the quinone imine dye MB, the xanthene dye RhB, the azo dye the azo dye X-3B, MO and RO5 was examined under similar conditions. As shown in Fig. 5(B), all the pollutants could also be decomposed efficiently. These results clearly indicated that the OMS-2/PMS is an efficient catalytic system for remediation of dyes wastewater.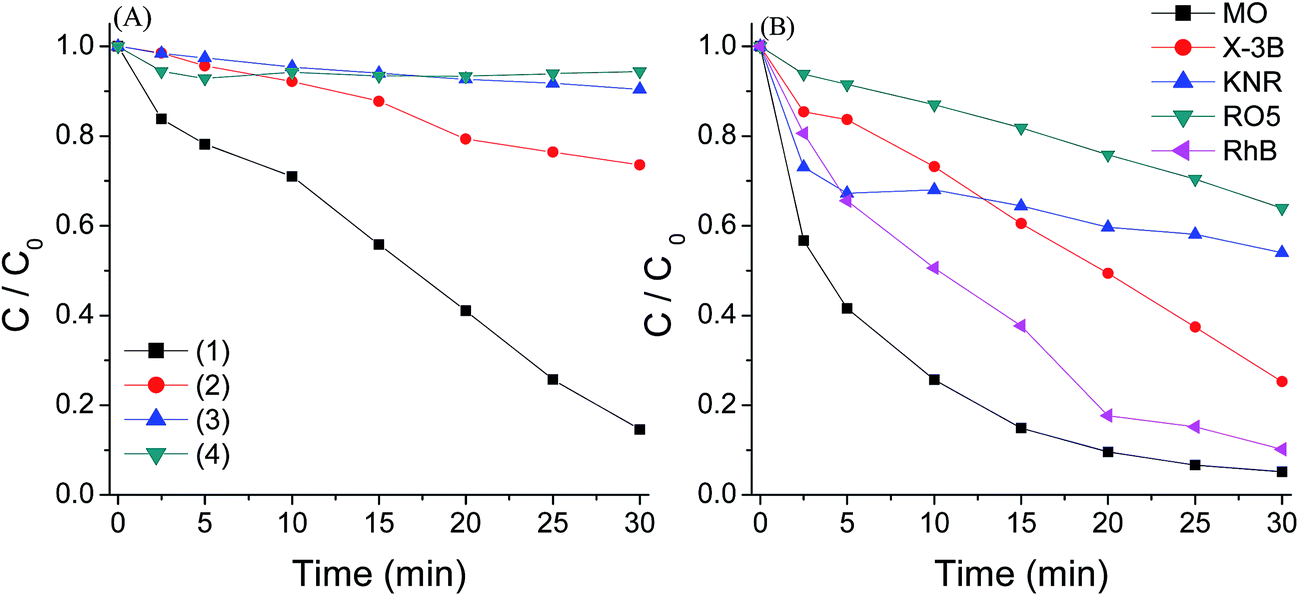 | ||
| Fig. 5 (A) AO7 degradation with different systems: (1) g-C3N4/PMS/Vis. (2) g-C3N4/Vis. (3) PMS/Vis. (4) g-C3N4/PMS. (B) Degradation of other organic dyes with g-C3N4/PMS/Vis system. Conditions: PMS 0.4 g L−1, g-C3N4 0.4 g L−1, dye 20 mg L−1, 25 °C. | ||
3.3. Intermediate products analysis
To provide further evidences of the effect of AO7 degradation with visible light irradiation in g-C3N4/PMS/Vis system, UV-vis and ESI-MS spectra were used to identify the degradation products. Representative UV-vis spectra changes observed during reaction are depicted in Fig. 6. For AO7 solution before reaction, it shows a main absorption bands at 484 nm, corresponding to the n–Π* transition of the azo form, and another band at 310 nm in ultraviolet region, which is attributed to the Π–Π* transition of the naphthalene ring.32 Addition of C3N4 and PMS into the aqueous solution caused the absorption bands of the dye in the visible region to decrease with time and finally to disappear, indicating the destruction of its chromophoric structure in the vicinity of the azo-linkage. At the same time, the decrease of the band in ultraviolet region was observed, due to the opening of the naphthalene ring. In addition, the absorbance at 254 nm first increased at the first 5 min and then descended gradually in the late reaction stage. The change can be attributed to the formation and degradation of a naphthalene type intermediates.14 Although AO7 was completely destructed within 30 min at 20 mg L−1 AO7, 0.4 g L−1 g-C3N4 and 0.40 g L−1 PMS, a very low TOC removal was observed in the system. This was probably caused by the low dosage of PMS and the short reaction time.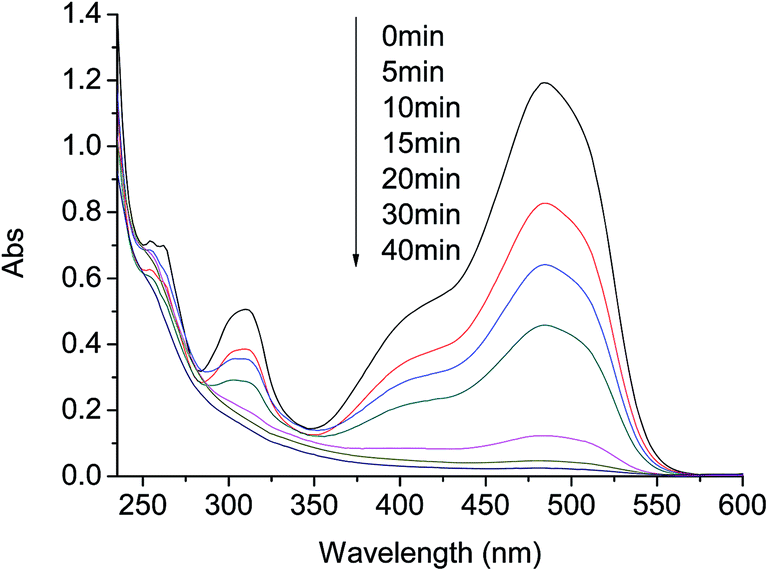 | ||
| Fig. 6 Representative UV-vis spectra changes during AO7 degradation by PMS/g-C3N4/Vis system. Conditions: PMS 0.4 g L−1, g-C3N4 0.4 g L−1, AO7 20 mg L−1, 25 °C. | ||
Fig. 7 shows the change of ESI-MS spectra of AO7 solution at positive and negative ion mode during reaction. In the ESI (+) mass spectra, at the beginning of the reaction, an intense ion of m/z 373 corresponding to [AO7 + Na]+ was observed as expected. After 20 min treatment, the intensity of AO7 at m/z 373 decreased significantly. Meanwhile, the peaks at m/z 103, 197, 213 and 251 showed up, indicating that it was degraded into some intermediate products. They could be attributed to 1,2-naphthaquinone and its further oxidized compounds.33 In the ESI (−) mass spectra, three degradation products, coming from the destruction of the azo groups and naphthalene rings, were identified: (1) 4-hydroxybenzenesulfonate (m/z 173), (2) 1,3-indanedione (m/z 145), according to the analysis of the fate of AOII degradation by other oxidation systems such as manganese peroxidase and UV/TiO2.34–36
 | ||
| Fig. 7 (A) ESI (+) and (B) ESI (−) mass spectra mass spectra of AO7 solution during degradation with g-C3N4/PMS/Vis system. Conditions: PMS 0.4 g L−1, g-C3N4 0.4 g L−1, AO7 20 mg L−1, 25 °C. | ||
3.4. Effect of operation parameters
To further illustrate the efficiency of g-C3N4/PMS/Vis system for AO7 degradation, the effect of various operation parameters including AO7 concentration, g-C3N4 concentration, PMS concentration, and solution pH was investigated. The influence of initial AO7 concentration at 10, 20, 30, 40 and 50 mg L−1 on the degradation rate is presented in Fig. 8. It can be seen that the degradation efficiency decreased with increasing AO7 concentration. At AO7 concentration of 10 mg L−1 near 86% removal was achieved in about 30 min, while at AO7 concentration of 50 mg L−1, the removal was 65% within 30 min. Due to the same concentrations of produced reactive radicals under the same concentrations of PMS and g-C3N4, high amount of dye in solution will require more time to achieve the same removal rate. However, the degradation rate did not decreased drastically at AO7 concentration of 20 mg L−1 compared with AO7 concentration of 10 mg L−1, probably owing to the fact that the g-C3N4/PMS/Vis system produced enough amounts of active radicals to remove low concentration of dye molecules under the experimental conditions. | ||
| Fig. 8 Influence of AO7 concentration on AO7 degradation with g-C3N4/PMS/Vis system. Conditions: PMS 0.4g L−1, g-C3N4 0.4 g L−1, 25 °C. | ||
Fig. 9 illustrates the AO7 removal with time at different catalyst dosages in solution. As expected, an enhancement of AO7 degradation was observed by increasing g-C3N4 concentration from 0.0 to 0.8 g L−1. A 10% removal of AO7 could be reached within 30 min at 0.0 g L−1 g-C3N4 loading. While complete removal could be reached within 20 min at g-C3N4 loading of 0.8 g L−1. The increased removal efficiency is evidently attributed to the increased availability of the active sites in the catalyst for reaction with PMS, which will generate more reactive radicals. At a higher g-C3N4 concentration of 1.2 g L−1, the removal rate of AO7 was higher than that at the concentration of 0.8 g L−1 during the first 5 min; but after the time, the rate became slightly lower, probably due to the increase of opacity of the suspension at a high catalyst concentration.
 | ||
| Fig. 9 Influence of g-C3N4 concentration on AO7 degradation with g-C3N4/PMS/Vis system. Conditions: PMS 0.4 g L−1, AO7 20 mg L−1, 25 °C. | ||
PMS concentration has a great impact on the treatment process efficiency and operational costs. To elucidate the role of PMS concentration on the degradation of AO7, some experiments were carried out by varying the initial PMS concentrations. As shown in Fig. 10, in the absence PMS, AO7 was slowly degraded with a removal of 26% after 30 min. After PMS was added to the aqueous solution with a concentration of 0.05 g L−1, near 39% extent of dye was removed. The incomplete removal of AO7 was probably due to lack of oxidant amount. When PMS concentration increased to 0.4 g L−1, AO7 was degraded faster with a removal of 86% after 30 min. The increasing rate of AO7 degradation with PMS increasing can be ascribed to more active radicals produced under a higher concentration of the oxidant. However, at a higher concentration of 0.8 g L−1, there was nearly no increase of the degradation rate of AO7. The PMS decomposition efficiency decreases as the PMS concentration increases. Thus, excessive oxidant cannot be used to degrade AO7 in the system.
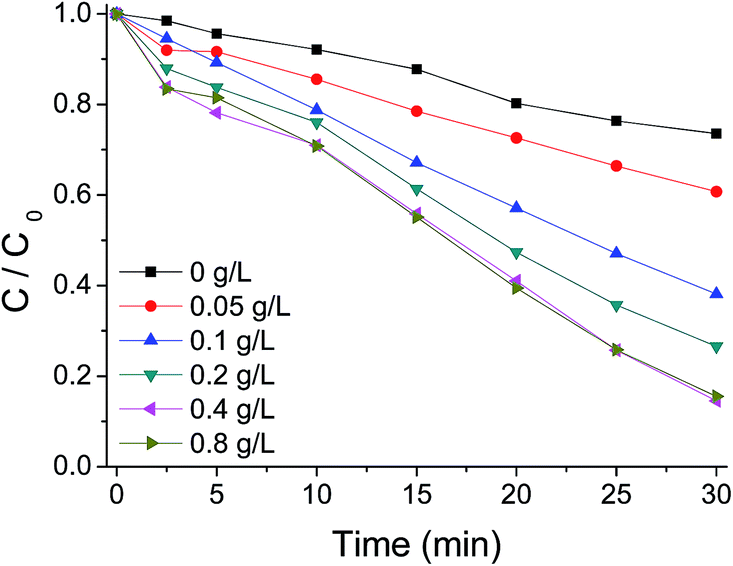 | ||
| Fig. 10 Influence of PMS concentration on AO7 degradation with g-C3N4/PMS/Vis system. Conditions: g-C3N4 0.4 g L−1, AO7 20 mg L−1, 25 °C. | ||
The influence of initial solution pH on AO7 degradation was also studied at five different pH values of 2.95, 4.19, 7.23, 9.59 and 11.84. As illustrated in Fig. 11, the degradation rate increased as pH increased from 2.95 to 11.84. In acidic medium, near 73% extent of dye was removed within 30 min. At neutral condition with pH of 7.23, the degradation rate became much faster with a final removal of 86%. When the oxidation was carried out at strong alkaline medium, the dye could be nearly complete degraded within 20 min. The lower rate of AO7 degradation at acidic medium might be attributed to the relatively higher stability of the oxidant at acidic pH values that could reduce the generation of radicals. Besides, in acidic condition, the formation of H-bond between H+ and the O–O group of HSO5− would be more significant that decrease the positive charge of HSO5−,37 thus hindering the interaction between HSO5− and the catalyst surface. Under alkaline conditions, the decomposition of PMS to active radicals will increase, which would enhance the degradation efficiency of target contaminant.
 | ||
| Fig. 11 Influence of initial solution pH on AO7 degradation with g-C3N4/PMS/Vis system. Conditions: PMS 0.4 g L−1, g-C3N4 0.4 g L−1, AO7 20 mg L−1, 25 °C. | ||
3.5. Stability of g-C3N4 catalyst
Stability is the most important character for a good solid catalyst in the solid–liquid reaction. To test the stability and recyclability of g-C3N4, the catalyst was collected by filtration after reaction, and the decolorization reaction was re-initiated by adding a fresh solution of AO7 and PMS. As shown in Fig. 12, the efficiency decreased during the second run in comparison with that in the first run, probably due to the reduction of adsorption ability. However, the catalyst was rather stable during the next runs, which could be used for six successive cycles with the removal of AO7 maintaining at about 75% after each run. The separated g-C3N4 catalyst after the sixth reaction run was examined by XRD and FT-IR with the results shown in Fig. 1(A) and (B). Compared with the images of the fresh catalyst, the XRD and FT-IR images of the recycled catalyst g-C3N4 did not show any obvious structural changes. From FT-IR spectra, it could be also seen that no externally adsorbed species detected on the catalyst surface. The XPS spectra of g-C3N4 were further measured after the six degradation experiments with the results shown in Fig. 4. In comparison with the images of the fresh catalyst, the XPS spectra of C1s and N1s images of the recycled catalyst also did not show any changes. The results indicated the excellent stability of the g-C3N4 catalyst in the presence of PMS with visible light irradiation.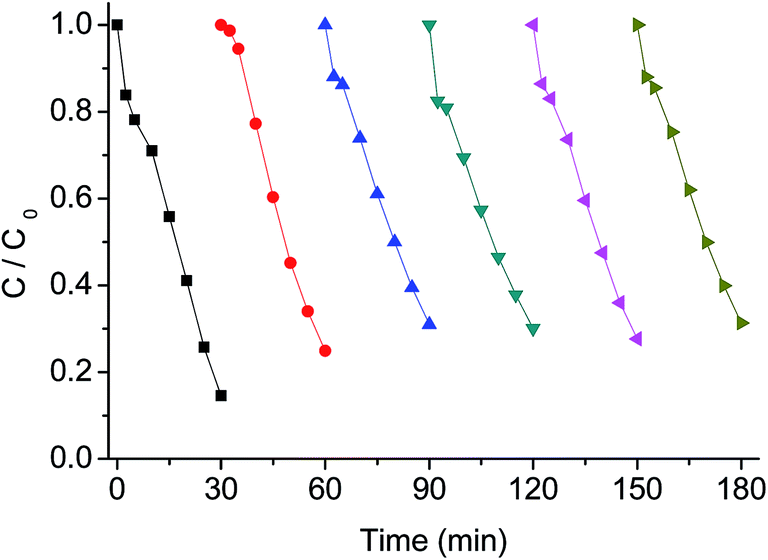 | ||
| Fig. 12 Degradation of AO7 using the recycled g-C3N4 catalyst. Condition: PMS 0.4g L−1, g-C3N4 0.4 g L−1, AO7 20 mg L−1, 25 °C. | ||
3.6. Activation mechanism of PMS on g-C3N4
PMS can be activated by UV, heat and catalysts to generate sulfate radical,38 which possess high oxidizing ability. HO˙ can be also generated by reaction of sulfate radical with H2O.39 Besides, the active species such as photo-generated electron (e−), photo-generated hole (h+) and superoxide radical (O2−˙) may also play a significant role in the photo-catalytic reaction. In order to understand the role of these active species in the g-C3N4/PMS/Vis system, quenching agents including EtOH, TBA, EDTA–2Na and 1.4-benzoquinone were added into the system. As shown in Fig. 13(A), in the presence of 1 mL TBA, a scavenger for hydroxyl radicals,40 the enhancement rather than inhibition of AO7 degradation was observed, indicating that HO˙ was not the main active species. It is known that the activation of oxidation occurs via an electron transfer. In the first step, the moderate oxidant O2−˙ is produced by the reaction of dissolved O2 with the first photoinduced electron and contributes to the degradation reaction. Thus, the increase of the decolorization in the presence of TBA may be due to the enhancement of O2−˙ radical stability in organic solvent. Indeed, the photocatalytic activity also was inhibited obviously by the addition of O2−˙ scavenger (BQ), further suggesting the presences of O2−˙ as the active species. The alcohols such as EtOH containing α-hydrogen can react with HO˙ and SO4−˙ radicals at significant rates.40 In the presence of 1 mL EtOH, the catalytic activity was obviously inhibited, implying that SO4−˙ radicals were another active species controlling the oxidation reaction, which can be produced from the reaction of PMS and e−. The adsorption and reaction of HSO5− in the surface of g-C3N4 catalyst can be proved by the XPS spectra of S2p for g-C3N4 after reaction. As indicated in Fig. 13(B), a strong peak at binding energy of 168.7 eV was observed. The peak more likely represents the signal from –SO3H group adsorbed by g-C3N4.41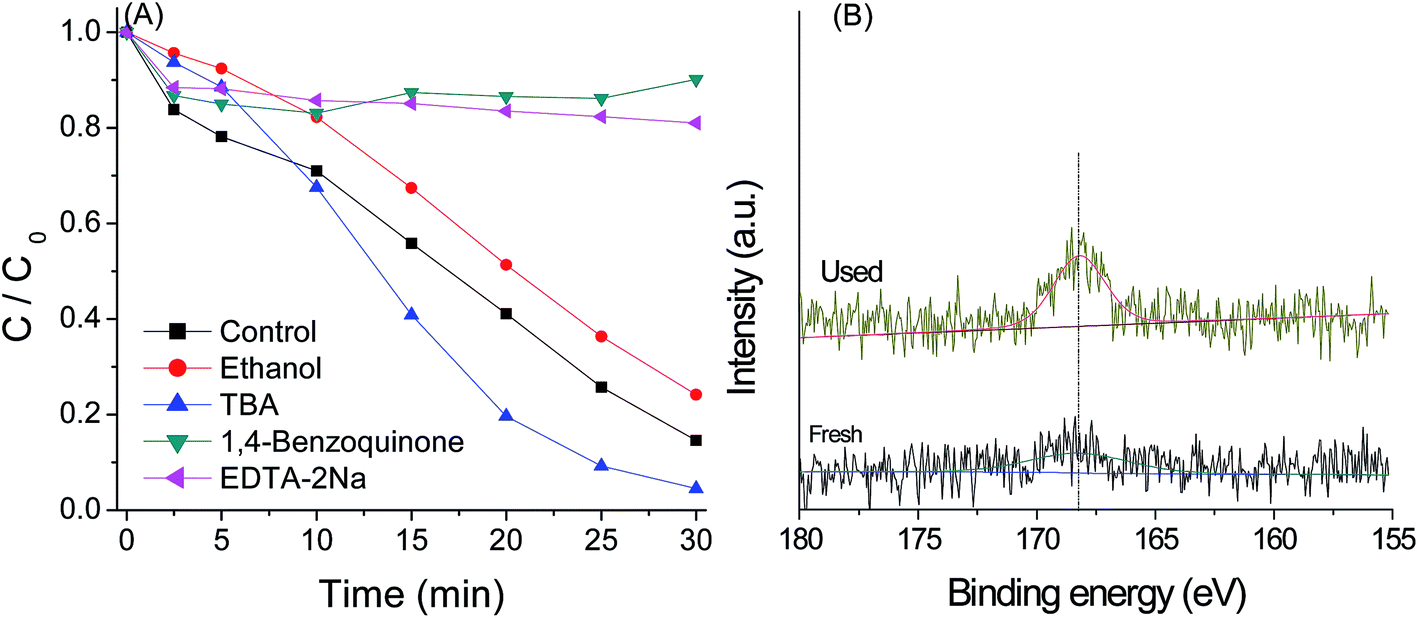 | ||
| Fig. 13 (A) Inhibiting effect of quenching agents on AO7 degradation with the g-C3N4/PMS/Vis system and (B) XPS spectra of S2p for g-C3N4 before and after reaction for the photocatalytic degradation of AO7 under visible light irradiation. Condition: PMS 0.4 g L−1, g-C3N4 0.4 g L−1, dye 20 mg L−1, 25 °C. | ||
It is reasonable that the valence band potential of g-C3N4 is too low to directly oxidize hydroxyl group to form hydroxyl radicals in thermodynamics,41 as confirmed by the addition of TBA. However, in the presence of 0.02 mmol EDTA-2Na, a kind of hole scavenger, the very low decolorization efficiency was observed. This might be due to that HSO5− react directly with the hole to produce SO5−˙, as the oxidant can be easily reduced and oxidized by many metal ions,14,15 which will increase the production of e−, and then SO4−˙ and O2−˙ radicals through the reaction between the oxidants and photoelectron. Similar conclusions have been drawn from the g-C3N4/H2O2/Vis system.19 According to these analyses, the mechanism of activation of PMS by g-C3N4 catalyst was proposed and is shown in Scheme 1, which mainly involved electron transfer under visible light irradiation, reduction of O2 and PMS by electron, oxidation of PMS by hole, and organic dyes degradation by SO4−˙ and O2−˙ radicals.
 | ||
| Scheme 1 The proposed mechanism of AO7 degradation in the g-C3N4/PMS/Vis system. | ||
4. Conclusions
The g-C3N4 catalyst was first used for heterogeneous activation of PMS and oxidative degradation of organic dyes in aqueous solution under visible light irradiation. Nearly complete removal of AO7 and other dyes could be achieved within 30 min on the catalyst. Increasing the AO7 concentration, catalyst amount and PMS concentration promoted the dye degradation, while with the rise of solution pH, the efficiency decreased. The catalyst also demonstrated stable performance in multiple runs. The quenching studies confirmed that sulfate radicals are the primary species produced during the catalytic decomposition of PMS. This photocatalytic process avoids the employment of any metal derivatives, and the catalyst was found to be stable and reusable, creating the promise of easily-available carbon nitride materials for environmental applications.Acknowledgements
This work was supported by the National Science Foundation of China (21304072 and 21207105) and the National Science & Technology Pillar Program (2014BAC13B02).References
- J. Herney-Ramirez, M. A. Vicente and L. M. Madeira, Appl. Catal., B, 2010, 98, 10 CrossRef CAS PubMed.
- F. Qi, W. Chu and B. Xu, Appl. Catal., B, 2013, 134–135, 324 CrossRef CAS PubMed.
- B. Legube and N. Karpel Vel Leitner, Catal. Today, 1999, 53, 61 CrossRef CAS.
- P. Shukla, H. Sun, S. Wang, H. M. Ang and M. O. Tadé, Sep. Purif. Technol., 2011, 77, 230 CrossRef CAS PubMed.
- I. S. X. Pinto, P. H. V. V. Pacheco, J. V. Coelho, E. Lorençon, J. D. Ardisson, J. D. Fabris, P. P. de Souza, K. W. H. Krambrock, L. C. A. Oliveira and M. C. Pereira, Appl. Catal., B, 2012, 119–120, 175 CrossRef CAS PubMed.
- J. Nawrocki and B. Kasprzyk-Hordern, Appl. Catal., B, 2010, 99, 27 CrossRef CAS PubMed.
- M. G. Antoniou, A. A. de la Cruz and D. D. Dionysiou, Appl. Catal., B, 2010, 96, 290 CrossRef CAS PubMed.
- S. Yang, P. Wang, X. Yang, L. Shan, W. Zhang, X. Shao and R. Niu, J. Hazard. Mater., 2010, 179, 552 CrossRef CAS PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Appl. Catal., B, 2004, 54, 155 CrossRef CAS PubMed.
- Y. R. Wang and W. Chu, Water Res., 2011, 45, 3883 CrossRef CAS PubMed.
- E. R. Bandala, M. A. Peláez, D. D. Dionysiou, S. Gelover, J. Garcia and D. Macías, J. Photochem. Photobiol., A, 2007, 186, 357 CrossRef CAS PubMed.
- K. H. Chan and W. Chu, Water Res., 2009, 43, 2513 CrossRef CAS PubMed.
- Y. Ding, L. Zhu, N. Wang and H. Tang, Appl. Catal., B, 2013, 129, 153 CrossRef CAS PubMed.
- L. Duan, B. Sun, M. Wei, S. Luo, F. Pan, A. Xu and X. Li, J. Hazard. Mater., 2015, 285, 356 CrossRef CAS PubMed.
- S. Luo, L. Duan, B. Sun, M. Wei, X. Li and A. Xu, Appl. Catal., B, 2015, 164, 92 CrossRef CAS PubMed.
- J. Cong, G. Wen, T. Huang, L. Deng and J. Ma, Chem. Eng. J., 2015, 264, 399 CrossRef CAS PubMed.
- J. Zhang, X. Shao, C. Shi and S. Yang, Chem. Eng. J., 2013, 232, 259 CrossRef CAS PubMed.
- H. Lee, H. Lee, J. Jeong, J. Lee, N. Park and C. Lee, Chem. Eng. J., 2015, 266, 28 CrossRef CAS PubMed.
- Y. Cui, Z. Ding, P. Liu, M. Antonietti, X. Fu and X. Wang, Phys. Chem. Chem. Phys., 2012, 14, 1455 RSC.
- Y. He, J. Cai, T. Li, Y. Wu, H. Lin, L. Zhao and M. Luo, Chem. Eng. J., 2013, 215–216, 721 CrossRef CAS PubMed.
- L. Ge, C. Han and J. Liu, Appl. Catal., B, 2011, 108–109, 100 CrossRef CAS PubMed.
- C. Han, L. Ge, C. Chen, Y. Li, X. Xiao, Y. Zhang and L. Guo, Appl. Catal., B, 2014, 147, 546 CrossRef CAS PubMed.
- L. Song, S. Zhang, X. Wu and Q. Wei, Chem. Eng. J., 2012, 184, 256 CrossRef CAS PubMed.
- C. Han, L. Wu, L. Ge, C. Y. Li and Z. Zhao, Carbon, 2015, 92, 31 CrossRef CAS PubMed.
- S. Huang, Y. Xu, M. Xie, H. Xu, M. He, J. Xia, L. Huang and H. Li, Colloids Surf., A, 2015, 478, 71 CrossRef CAS PubMed.
- H. Zhai, L. Cao and X. Xia, Chin. Chem. Lett., 2013, 24, 103 CrossRef CAS PubMed.
- X. Zhang, J. Hu and H. Jiang, Chem. Eng. J., 2014, 256, 230 CrossRef CAS PubMed.
- H. Qian, H. Huang and X. Wang, J. Power Sources, 2015, 275, 734 CrossRef CAS PubMed.
- S. Wu, Y. Yu and W. Zhang, Mater. Sci. Semicond. Process., 2014, 24, 15 CrossRef CAS PubMed.
- M. B. Ansari, H. Jin, M. N. Parvin and S. Park, Catal. Today, 2012, 185, 211 CrossRef CAS PubMed.
- H. Ji, F. Chang, X. Hu, W. Qin and J. Shen, Chem. Eng. J., 2013, 218, 183 CrossRef CAS PubMed.
- M. Luo, L. Lv, G. Deng, W. Yao, Y. Ruan, X. Li and A. Xu, Appl. Catal., A, 2014, 469, 198 CrossRef CAS PubMed.
- P. Muthirulan, C. N. Devi and M. M. Sundaram, Ceram. Int., 2014, 40, 5945 CrossRef CAS PubMed.
- X. Chen, W. Wang, H. Xiao, C. Hong, F. Zhu, Y. Yao and Z. Xue, Chem. Eng. J., 2012, 193–194, 290 CrossRef CAS PubMed.
- C. Cai, H. Zhang, X. Zhong and L. Hou, Water Res., 2014, 66, 473 CrossRef CAS PubMed.
- M. Stylidi, D. I. Kondarides and X. E. Verykios, Appl. Catal., B, 2003, 40, 271 CrossRef CAS.
- J. Liu, Z. Zhao, P. Shao and F. Cui, Chem. Eng. J., 2015, 262, 854 CrossRef CAS PubMed.
- Y. Guan, J. Ma, Y. Ren, Y. Liu, J. Xiao, L. Lin and C. Zhang, Water Res., 2013, 47, 5431 CrossRef CAS PubMed.
- L. J. Xu, W. Chu and L. Gan, Chem. Eng. J., 2015, 263, 435 CrossRef CAS PubMed.
- W. Shi, Q. Cheng, P. Zhang, Y. Ding, H. Dong, L. Duan, X. Li and A. Xu, Catal. Commun., 2014, 56, 32 CrossRef CAS PubMed.
- L. Zhang, D. Liu, J. Guan, X. Chen, X. Guo, F. Zhao, T. Hou and X. Mu, Mater. Res. Bull., 2014, 59, 84 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |