
Injectable supramolecular hydrogels fabricated from PEGylated doxorubicin prodrug and α-cyclodextrin for pH-triggered drug delivery†
Abstract
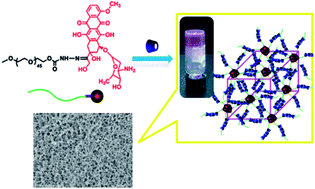
Supramolecular hydrogels, which are held together by noncovalent bonds and show responses to external stimuli, are of great interest in therapeutic delivery and tissue engineering as the injectable depot systems. To obtain a supramolecular hydrogel with multifunctions, such as low cytotoxicity, injectability and stimuli-triggered drug release, we herein report on the synthesis and characterization of a supramolecular hydrogel, which was formed by host–guest interaction between α-cyclodextrin (α-CD) and a PEGylated doxorubicin prodrug linked with an acid-cleavable hydrazone group (mPEG-Hyd-DOX). The polymeric prodrug displayed lower cytotoxicity than the free DOX. The host–guest interaction was demonstrated by X-ray diffraction (XRD) analysis. The structures and morphologies of the supramolecular hydrogels were systematically investigated by differential scanning calorimetry (DSC), scanning electron microscopy (SEM) and thermogravimetric analysis (TGA). The sol–gel transition process was monitored by dynamic and steady rheological analysis. The hydrogels could be degraded in the acidic environment of tumor cells and achieved the controlled delivery of DOX. The results of the pH-responsive property, in vitro cytotoxicity and drug release revealed that the supramolecular hydrogels can be used as a potential injectable matrix for the encapsulation and controlled release of anticancer drugs. This study provides an alternative for the construction of dual- or multi-drug delivery systems.
 Please wait while we load your content...
Please wait while we load your content...

