
Single-source mediated facile electrosynthesis of p-Cu2S thin films on TCO (SnO2:F) with enhanced photocatalytic activities†
Abstract
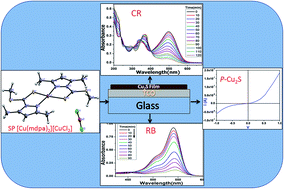
Electrosynthesis of p-Cu2S thin films on a fluorine-doped tin oxide coated transparent conducting TCO (SnO2:F) glass substrate is carried out by chronoamperometry and cyclic voltammetry (CV) using an ethanolic solution of a single-source precursor (SP), [Cu(mdpa)2][CuCl2] (where mdpa is 3,5-dimethyl pyrazole-1-dithioic acid). The appropriate potential at which the formation of stoichiometric p-Cu2S thin films occurs was found to be −0.48 V. The mechanism of the selective deposition of the p-Cu2S phase can be described by the electroreduction of Cu–N/S bonds in the coordination sphere following the dissociation of a precursor complex into Cu+ and mdpa. The free ligand mdpa is reduced to sulfide ion producing volatile organics in the electrochemical process. The quality deposition of thin films depends on the optimization of the SP concentration. An X-ray diffraction study reveals the high chalcosite phase of copper sulfide with preferential orientation along the (110) plane. The I–V characteristic of the as deposited Cu2S/TCO thin film shows a non-ohmic behavior suggesting the formation of a p–n heterojunction diode. The p-Cu2S/TCO thin films are found to be excellent photocatalysts for the photo-degradation of Congo Red (CR) under visible light irradiation. It has also been shown that the photocatalytic activity of the deposited thin films increased many fold with the addition of a catalytic amount of hydrogen peroxide in the photo-degradation of Rose Bengal (RB) dye under visible light irradiation. A possible mechanism for the improved photoactivity of p-Cu2S/TCO is proposed and involves the electron scavenging property of H2O2 followed by OH− radical formation, significantly accelerating the photodegradation of RB dye.
 Please wait while we load your content...
Please wait while we load your content...

