
Development and validation of kinetic and atomic absorption spectrophotometric methods for the determination of salbutamol sulfate
R'afat Mahmoud Nejem*a,
Mahmoud Mohamed Issab,
Mozer Hosny AL-Kholyb,
Akila Amin Salehc,
Alaa Abu Shanabd,
Raluca Ioana Stefan van Stadene and
Hassan Y. Aboul-Eneinf
aAnalytical Chemistry, Department of Chemistry, Alaqsa University, P. O. Box 4051, Gaza, Palestine. E-mail: rafat_nejem@yahoo.com; Fax: +970 282845488; Tel: +970 599603459
bPharmaceutical Analytical Chemistry, Department of Chemistry, Alaqsa University, P. O. Box 4051, Gaza, Palestine
cInorganic Chemistry, Ain Shams University, Cairo, Egypt
dInorganic Analytical Chemistry, Department of Chemistry, Alaqsa University, P. O. Box 4051 Gaza, Palestine
eNational Institute of Research for Electrochemistry and Condensed Matter, Bucharest, Romania
fPharmaceutical and Medicinal Chemistry Department, Pharmaceutical and Drug Industries Research Division, National Research Center, Dokki, Giza 12622, Egypt. E-mail: haboulenein@yahoo.com; Fax: +20 233370931; Tel: +20 1003678948
First published on 5th June 2015
Abstract
Two sensitive kinetic and atomic absorption spectrophotometric (AAS) methods were developed for the determination of salbutamol sulfate (SLS) in its dosage forms. The kinetic method was based on the bromination reaction of the drug by bromine generated in situ by the reaction of bromate with bromide in acidic medium. The reaction was followed spectrophotometrically by measuring the decrease of bromine color at 380 nm. The reaction was carefully studied and optimized. Under optimum conditions, the stoichiometry and the order of the reaction were determined. The initial rate and fixed time methods were utilized for the determination of salbutamol sulfate concentrations. The AAS method depended on the oxidation of iron(II) with excess bromine from the bromination reaction of the drug. A new method for separation of iron(III) was used. Then iron(II) in the aqueous layer was aspirated into an air–acetylene flame and determined by AAS. The linear ranges for the proposed methods were 2.0–10.0 and 0.2–2.0 μg mL−1 with detection limit of 0.30 and 0.012 μg mL−1 for kinetic and AAS, respectively. The proposed methods were validated; the mean recovery ranges from 98.0 to 102.0% with RSD < 2.1%. Common excipients did not interfere with the measurements of SLS. The methods were successfully applied to determine SLS in dosage forms; there was no significant difference between the proposed methods and official one.
1. Introduction
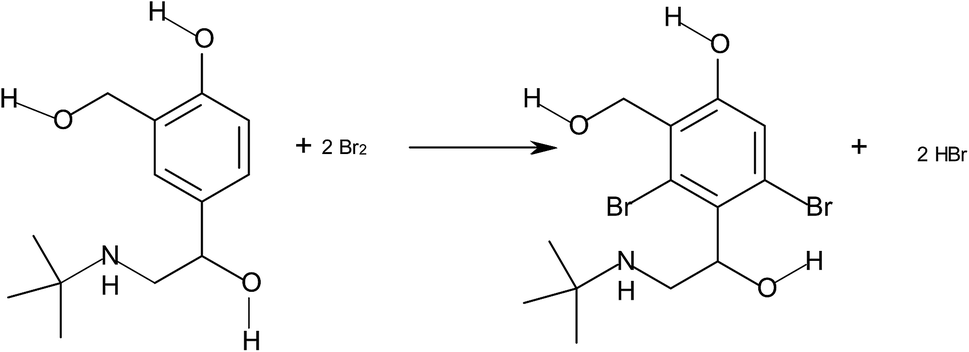
Asthma is a chronic respiratory disease characterized by inflammation and narrowing of airways in the lungs, the bronchi. During an asthma attack, the smooth muscle surrounding the bronchi contracts and the lining of the bronchi swells; this swelling is life-threatening because the airways can become blocked. Salbutamol is one of the β-agonist bronchodilators, the largest group among the various classes of inhaled asthma drugs. Salbutamol sulfate, α-1-[[(1,1-dimethylethyl)amino]methyl]-4-hydroxy-1,3-benzenedimethanol sulfate (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Scheme 1) is a direct-acting sympathomimetic agent with a relatively selective action on β2-adrenoacceptors. The clinical use of salbutamol is the management of reversible airways observation such that occurs in asthma and delaying premature labour.1
:1) (Scheme 1) is a direct-acting sympathomimetic agent with a relatively selective action on β2-adrenoacceptors. The clinical use of salbutamol is the management of reversible airways observation such that occurs in asthma and delaying premature labour.1
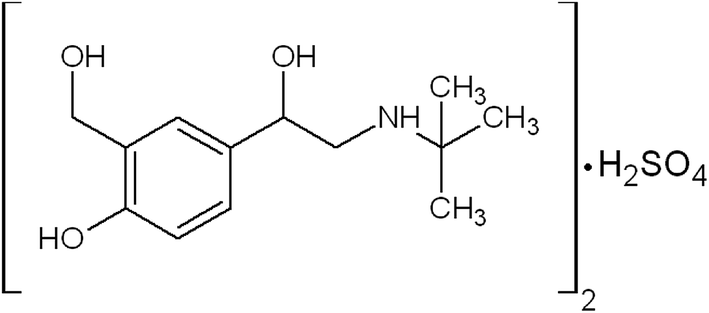 | ||
| Scheme 1 Chemical structure of salbutamol sulfate. | ||
Literature survey shows that several spectrophotometric,2–12 spectrofluorimetric,13 conductometric,14 potentiometric,15 LC-MS16 and HPLC17–23 methods reported for the determination of salbutamol sulfate in various samples. So far, only one kinetic spectrophotometric method has been reported for determination of salbutamol sulfate.24 No atomic absorption spectroscopic method has been reported for determination of salbutamol sulfate. Basavaiah and Prameela used the bleaching of methyl orange for the kinetic bromination of salbutamol but nothing is mentioned in the paper about the different kinetic behavior for the simultaneous reaction of bromine with salbutamol sulfate and methyl orange.24
In this paper, bromine is yielded in a slow reaction according to the following stoichiometry:
| BrO3− + 5Br− + 6H+ → 3Br2 + 3H2O |
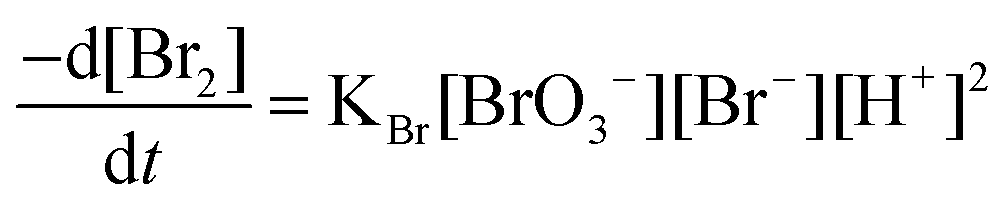
The kinetic method depends on the bromination of the drug by bromine generated in situ by the interaction of bromate with bromide in acidic medium. The absorbance of bromine color decreases with time and hence, a kinetically based method is elaborated. The use of bromine in kinetic spectrophotometric method is becoming of great interest26–28 as it offered some advantages:
•Bromine can act in different reaction ways such as: substitution, addition and oxidation.
•Reaction between bromine and salbutamol sulfate is slow.
•Bromine can be detected in both UV and visible absorption bands.
The atomic absorption method is based on the oxidation of iron(II) with unreacted bromine from the bromination reaction of the drug. A new method for the separation of iron(III) with ammonium thiocyanate/n-propyl alcohol extraction system in the presence of sodium chloride has been used.29 Then the remained iron(II) in aqueous layer is aspirated into air–acetylene flame and determined by atomic absorption spectrophotometer.
The proposed methods have the advantages of being simple, sensitive and free from interferences.
2. Results and discussion
In acidic medium, bromate oxidizes bromide to bromine and the in situ generated bromine reacts with the drug. The absorbance of the unreacted bromine decreases with time and hence, a kinetically based method was elaborated (Fig. 1). A study of optimum conditions was carried out and stoichiometry of the reaction of salbutamol with bromine was ascertained.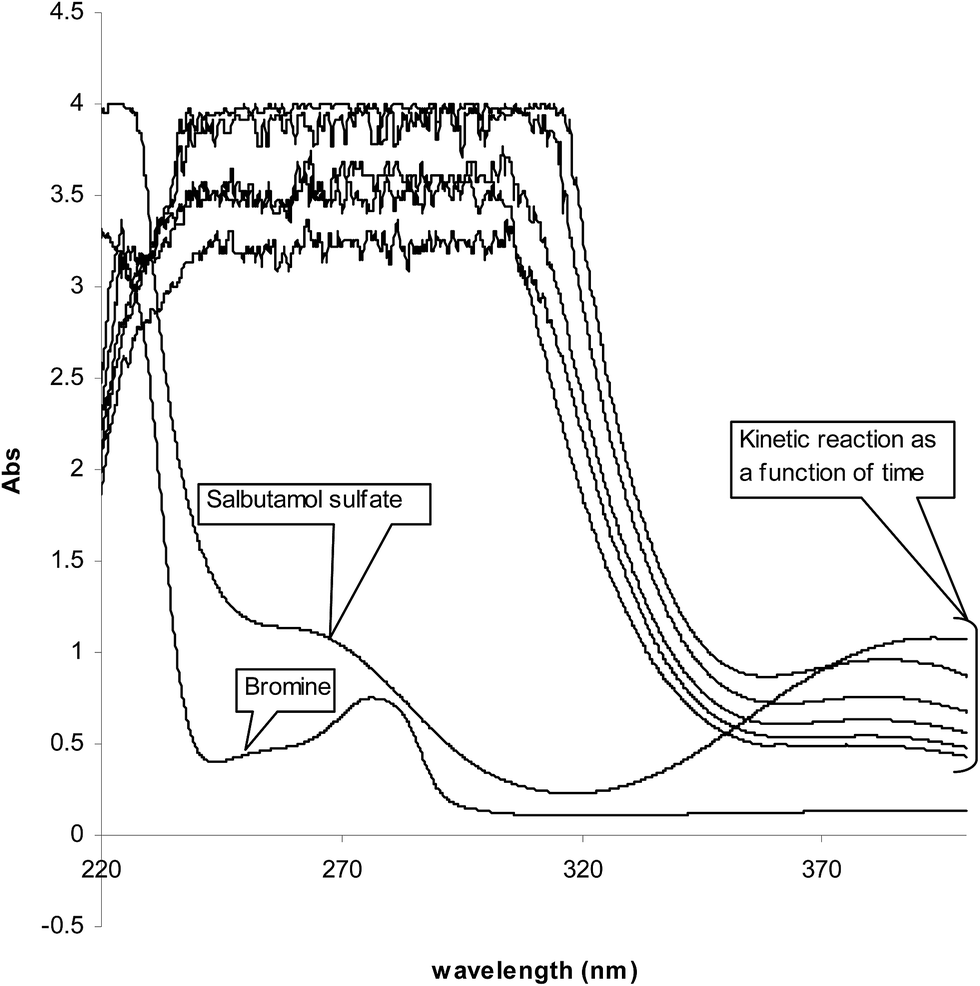 | ||
| Fig. 1 Absorption spectra of (1) bromine, 1.8 × 10−3; (2) SLS, 3.0 × 10−5 mol L−1; (3) kinetic reaction as a function of time at 10 °C. | ||
The pseudo-order rate constant (K′) was determined by studying the reaction at different temperatures; 5, 10, 15, 20 and 25 °C using fixed concentration of SLS and Br2 (Table 1). It was found that the rate of reaction affected by the change of temperature. The rate of the reaction at 10 °C was moderate, so that it was possible to estimate their reaction rate by fitting the kinetic data using a suitable regression method. Furthermore, the rates of reactions at 15, 20, 25 °C were so fast that was not possible to determine their rate constants.
| T (°C) |
|
Log[SLS] | K′ S−1 |
|---|---|---|---|
| a ND: not detected. | |||
| 10 | −3.478 | −5.22 | 46.23 |
| 10 | −3.176 | −4.92 | 46.23 |
| 10 | −3.000 | −4.74 | 46.23 |
| 10 | −2.875 | −4.62 | 46.23 |
| 10 | −2.789 | −4.52 | 46.23 |
| 5 | −3.988 | −5.22 | 15.50 |
| 15 | ND | −5.22 | ND |
| 20 | ND | −5.22 | ND |
| 25 | ND | −5.22 | ND |
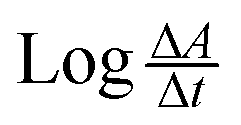
No change in the reaction stoichiometry was occurred when 0.5–2.0 mL of 5 mol L−1 H2SO4 acid were used. So, a 1.0 mL of 5 mol L−1 H2SO4 in a total volume of 10.0 mL was found suitable for bromination of SLS drug.
The bromide concentration was in large excess (0.1 mol L−1) in order to accelerate bromine generation. The amount of generated bromine was dictated by bromate.
An initial concentration of 3 × 10−4 mol L−1 bromate generated 9.0 × 10−4 mol L−1 bromine. This was a suitable concentration for experimental purpose. Fig. 2 illustrates some reaction profiles of the bromine consumption, followed at 380 nm, for different initial bromine concentration when [SLS] > [Br2]. Fig. 3 explains the bromine consumption for different salbutamol sulfate concentrations when [Br2] > [SLS].
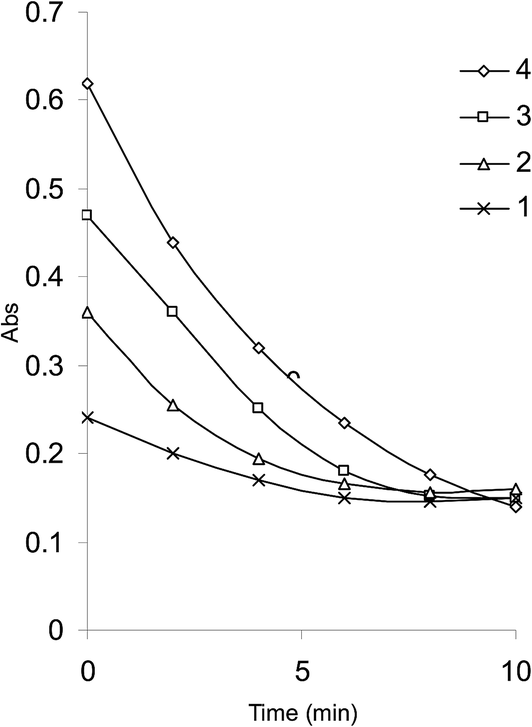 | ||
| Fig. 2 Bromine consumption in the SLS–Br2 system for different initial bromine concentrations. [Br2] (1) 4.5 × 10−4; (2) 6.75 × 10−4; (3) 9.0 × 10−4; (4) 1.13 × 10−3 mol L−1, [SLS] 3 × 10−3 mol L−1; H2SO4 0.5 mol L−1. | ||
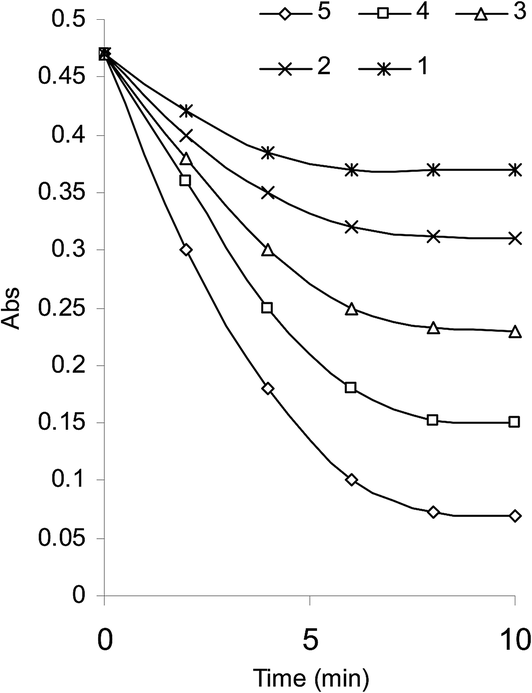 | ||
| Fig. 3 Bromine consumption in the SLS–Br2 system for different SLS concentrations (1) 5.97 × 10−6; (2) 1.19 × 10−5; (3) 1.79 × 10−5; (4) 2.39 × 10−5; (5) 2.99 × 10−5 mol L−1, [Br2] 9 × 10−4 mol L−1; H2SO4 0.5 mol L−1. | ||
The stoichiometry of the reaction was established from limiting logarithms plot in Fig. 4. The plots of log absorbance vs. log[Br2] and [salbutamol] exhibit slope values of 1.85 and 0.99, respectively. Hence, it is concluded that the reaction proceeds at the molar ratio of 2:1. The probable reaction is:
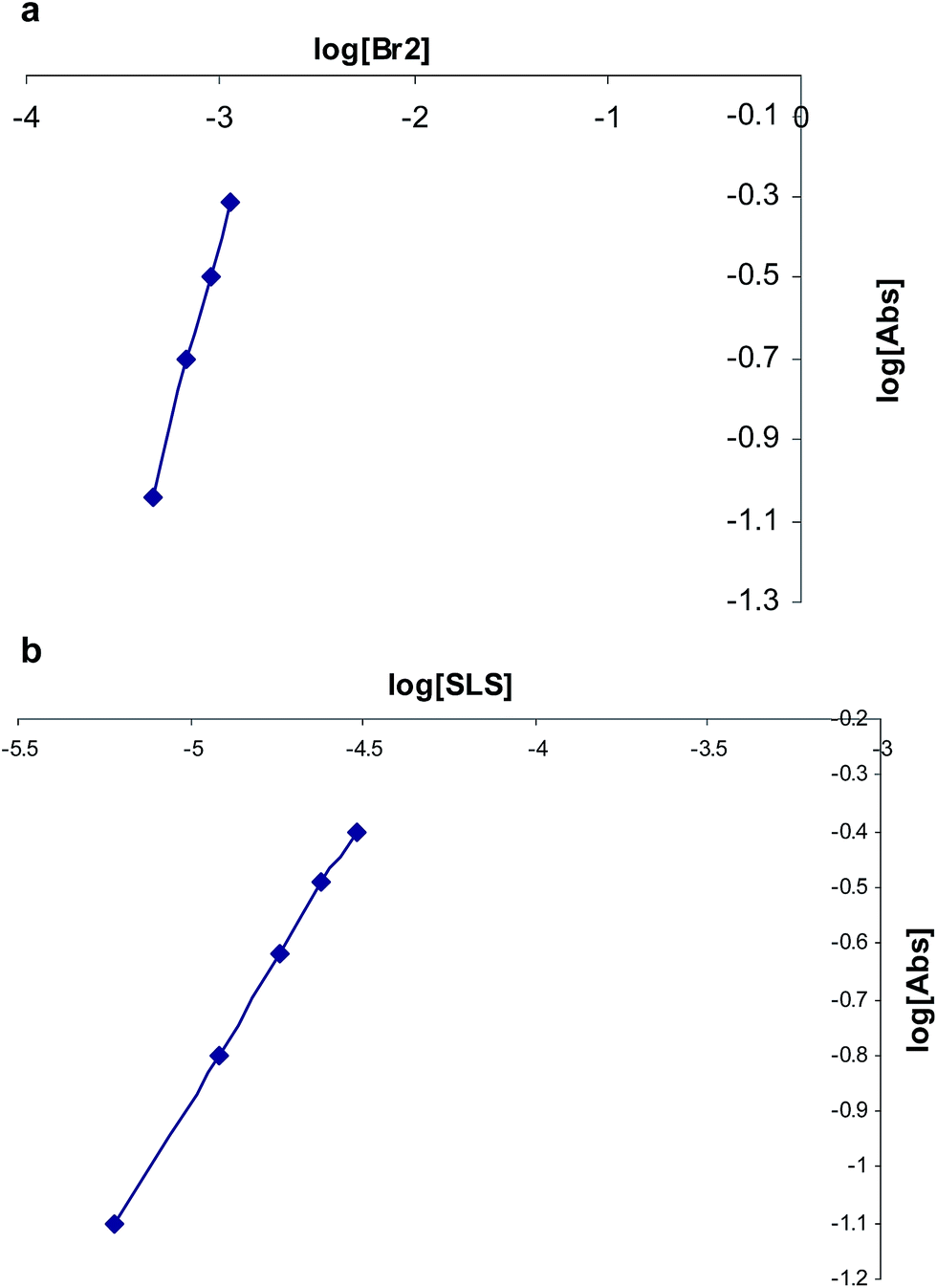 | ||
| Fig. 4 Limiting logarithmic plots for the molar ratio. (a) Log[Abs] vs. log[Br2] and (b) log[Abs] vs. log[SLS]. | ||
The initial-rate and fixed-time methods were adopted for constructing the calibration curves. The initial absorbance versus time is shown in Fig. 3. The rate expression can be written as:
| rate = K[Br2]m[SLS]n |
| rate = K′[SLS]n |
| Lograte = logK′ + nlogC |
The rate of the reaction may be estimated as 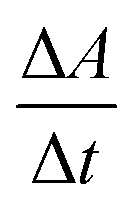 , where A is the absorbance and t is the time in seconds. By compensating the logarithm of rates and concentrations of SLS, as shown in Table 1, in the above equation, consequently:
, where A is the absorbance and t is the time in seconds. By compensating the logarithm of rates and concentrations of SLS, as shown in Table 1, in the above equation, consequently:
| Lograte = 1.665 + 0.983logC (r = 0.9987) |
Therefore, K′ = 46.23 S−1 and the reaction is first order [n = 0.983] with respect to salbutamol sulfate. The calibration graph showed a linear relationship over the concentration rang 5.97 × 10−6–2.99 × 10−5 mol L−1 (2–10 μg mL−1). The low value of detection limit 8.90 × 10−7 mol L−1 (0.3 μg mL−1) confirmed the good sensitivity of the proposed kinetic method.
Regression equation for salbutamol sulfate at different fixed time over the range of 5.97 × 10−6–2.99 × 10−5 mol L−1 (2–10 μg mL−1) at 380 nm is shown in Table 2. It is clear that the most acceptable values of the correlation coefficient and the intercept were obtained for a fixed time 10.0 min, which was therefore chosen as the most suitable time interval for measurement. After optimizing the reaction conditions, the fixed time methods was applied to the determination of salbutamol sulfate over the range 2–10 μg mL−1 using the following equation:
| A10 = 0.002 + 0.0407C (r = 0.9990) |
| Time (min) | Regression equation | r | LOD (μg mL−1) |
|---|---|---|---|
| 2.5 | A = −0.0020 + 0.0221C | 0.9820 | 1.23 |
| 5.0 | A = −0.0015 + 0.0294C | 0.9840 | 0.60 |
| 7.5 | A = 0.0010 + 0.0393C | 0.9953 | 0.44 |
| 10 | A = 0.0020 + 0.0407C | 0.9990 | 0.29 |
In atomic absorption spectrophotmetric method, the unreacted bromine is reduced by iron(II) and the resulting iron(III) complexed with thiocyanate. Then, the complex is extracted and iron(II) in aqueous layer determined by the proposed AAS. As a result there is a proportional increase in the absorbance of iron(II) with the increasing concentration of SLS. A 1.0 mL of 20 μg mL−1 iron(II) in a total volume of 10 mL resulted in convenient absorbance. This was found to be completely reduced by 10 μg bromate in the presence of excess bromide. Therefore, different amounts of SLS were reacted with 10 μg bromate (each 1.0 μg SLS reacted with 0.333 μg bromate). An overall acidity of 0.5 mol L−1 sulfuric acid used for the bromination between SLS and bromate was also maintained for reducing step. The bromination reaction was found to be complete in 10 min (fixed – time method) for the 0.2–2.0 μg mL−1 ranges of SLS studied and a contact time of 10 min was found necessary for reducing iron(II). After optimizing the conditions, the AAS method was applied to the determination of SLS over the range 0.2–2.0 μg mL−1using the following equation:
| A = −0.032 + 0.518C (r = 0.9982) |
2.1 Analytical data
The analytical characteristics of the three proposed method are shown in Tables 3–5. Initial rate method was found to be applicable over the range 5.97 × 10−6–2.99 × 10−5 mol L−1 (2.0–10.0 μg mL−1) under the described experimental conditions. The limit of detection (LOD) was calculated and found to be 8.90 × 10−7 mol L−1 (0.3 μg mL−1) as shown in Table 3. In the fixed–time method, the absorbance of the reaction solution containing varying amounts of SLS was measured at a preselected fixed time. Beer's law was obeyed in the concentration range 2.0–10.0 μg mL−1. The LOD value was 0.31 μg mL−1 as in Table 4. The low value confirmed the good sensitivity of the fixed time method. The linear plot gave regression equation:| A10 = 0.002 + 0.0407C (r = 0.9990) |
| Conc. (μg mL−1) | Intraday assay (LOD = 0.30 μg mL−1) | ||||
|---|---|---|---|---|---|
| Found ± SDa | Recovery, % | SAEb | RSD % | CLc | |
| a Average of five determinations.b SAE, standard analytical error.c 95% confidence limits (n = 5). | |||||
| 2.0 | 1.98 ± 0.02 | 99.00 | 0.009 | 1.010 | 1.98 ± 0.024 |
| 6.0 | 6.10 ± 0.06 | 101.7 | 0.026 | 0.980 | 6.10 ± 0.072 |
| 10.0 | 9.80 ± 0.09 | 98.00 | 0.040 | 0.920 | 9.80 ± 0.111 |
| Conc. (μg mL−1) | Intraday assay (LOD = 0.31 μg mL−1) | ||||
|---|---|---|---|---|---|
| Found ± SDa | Recovery, % | SAEb | RSD % | CLc | |
| a Average of five determinations.b SAE, standard analytical error.c 95% confidence limits (n = 5). | |||||
| 2.0 | 1.99 ± 0.018 | 99.50 | 0.008 | 0.90 | 1.99 ± 0.022 |
| 6.0 | 5.98 ± 0.05 | 99.60 | 0.022 | 0.83 | 5.98 ± 0.062 |
| 10.0 | 9.86 ± 0.16 | 98.60 | 0.071 | 1.62 | 9.86 ± 0.19 |
The calibration graph for AAS procedure was obtained by plotting the absorbance of iron(II) remain in aqueous layer with the concentration of SLS. Linearity was observed in the range 0.2–2.0 μg mL−1 and could be described by the recession equation:
| A = −0.032 + 0.518C (r = 0.9982) |
The LOD was calculated and found to be 0.026 μg mL−1 (Table 5). Based on the sensitivity results obtained, the AAS method has the superiority of over the other two methods.
| Conc. (μg mL−1) | Intraday assay (LOD = 0.026 μg mL−1) | ||||
|---|---|---|---|---|---|
| Found ± SDa | Recovery, % | SAEb | RSD % | CLc | |
| a Average of five determinations.b SAE, standard analytical error.c 95% confidence limits (n = 5). | |||||
| 0.4 | 0.393 ± 0.007 | 98.80 | 0.003 | 1.80 | 0.393 ± 0.008 |
| 1.2 | 1.23 ± 0.026 | 100.2 | 0.012 | 2.10 | 1.23 ± 0.032 |
| 2.0 | 1.990 ± 0.042 | 99.50 | 0.019 | 2.10 | 1.990 ± 0.077 |
2.2 Precision and accuracy
The intraday and interday precisions for the three methods were found to be in the range 0.83–3.81% and 0.9–3.5%, respectively (Tables 3–5). These data indicated that the proposed methods were reproducible within and between days.The percent errors were found to vary from 1.0–2.0%. The mean percentage recovery ranged from 98.0–102.5% with relative standard deviation values < 4%. The percent errors and the RSD values indicate the high accuracy and precision of the methods.
2.3 Interferences
The selectivity of the proposed methods was preliminarily checked by determining of SLS in the presence of other compounds of the tablet. The results are summarized in Table 6. It is evident that the excipients had no effect on the SLS estimation. Hence, the determination of the drug by the proposed methods is considered to be free from interference due to excipients.| Initial-rate method | Fixed-time method | AAS method | ||||||
|---|---|---|---|---|---|---|---|---|
| Taken (μg mL−1) | Found (μg mL−1) | Recoveryb, % | Taken (μg mL−1) | Found (μg mL−1) | Recovery, % | Taken (μg mL−1) | Found (μg mL−1) | Recovery, % |
| a Synthetic mixture containing excipients (starch (30 mg), talc (2 mg), lactose (140 mg), magnesium stearate (2 mg) and croscarmellose sodium (2 mg)) in water.b Mean of five determination. | ||||||||
| 2.0 | 2.04 | 102.0 | 2.0 | 1.98 | 99.00 | 0.4 | 0.39 | 98.00 |
| 6.0 | 5.88 | 98.00 | 6.0 | 5.90 | 98.30 | 1.2 | 1.22 | 101.6 |
| 10.0 | 9.86 | 98.60 | 10.0 | 10.1 | 101.0 | 2.0 | 2.02 | 101.0 |
2.4 Application to dosage forms
The proposed methods has been successful applied to the determination of SLS and the results were statistically compared with those obtained by use the official methods.31 The results in Table 7 show that there is no significant difference between the performances of the methods compared.3. Experimental
3.1 Apparatus
A shimadzu (Kyoto, Japan) UV-1650 PC, UV-Visible double-beam spectrophotometer with two matched 1 cm path-length quartz cells was used for the measurements of molecular absorption. The subsequent statistical manipulation was performed by transferring the spectral data to Microsoft Excel 2007 program and processing them with the standard curve fit package and matrix calculation.Flame atomic absorption measurements were performed using a Perkins-Elmer AAS, Model A, Analyst 100 spectrophotometer equipped with an iron hollow-cathode lamp, under the following conditions: 302.1 nm wavelength, 30.0 mA lamp current, 0.2 nm slit width and 3.5:1.5 air:acetylene ratio.
3.2 Reagents and solutions
All reagents used were of analytical reagent grade and double-distilled water was used for the preparation of solutions. Pharmaceutical grade salbutamol sulfate was kindly provided by the Middle East pharmaceuticals and cosmetics laboratories, Palestine. Stock solution of 5.97 × 10−3 mol L−1 salbutamol sulfate was prepared daily by dissolving 0.20 g SLS in 100 mL of double-distilled water.Ferrous ammonium sulfate of 3.57 × 10−2 mol L−1 was prepared by dissolving 1.4 g the solid salt in 100 mL of water. A standard solution of 9 × 10−3 mol L−1 potassium bromate was prepared by dissolving 0.15 g KBrO3 in 100 mL of water. All required diluted working solutions were prepared by addition of double-distilled water.
A 10% potassium bromide, 5.0 mol L−1 sulfuric acid and 1.0 mol L−1 ammonium thiocyanate were also prepared.
3.3 Analytical procedures
where b is the slope and S is the standard deviation of the regression line. Three concentrations where selected and five solutions of each were used for intraday and interday analysis. Recovery experiments were performed using the standard addition method. The standard analytical error (SAE) and 95% confidence limits (CL) were calculated as:
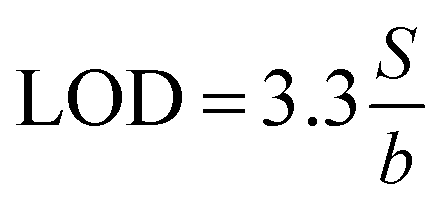
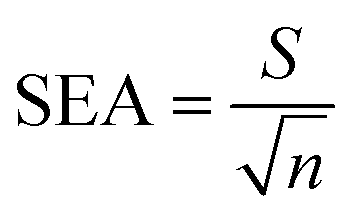
where S is the standard deviation of n measurements and t is the tabulated t-value.
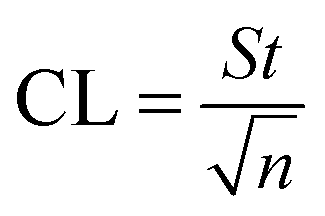
4. Conclusion
The proposed methods described in this paper are simple, selective, sensitive and reproducible and don't require expensive reagents and sophisticated instruments. These methods are applicable for routine analysis of the studied drug in raw materials and pharmaceutical formulations over wide concentration range without interference from common excipients. The methods can use both spectrophotometric and (AAS) techniques for the final measurement step. The statistical parameters indicate the reproducibility and accuracy of the methods.Sensitive kinetic and AAS methods are developed, to determine SLS in its dosage forms. The initial rate and fixed time methods are used for the determination of SLS in kinetic method and a new way for separation iron(III) was used in AAS methods.
The AAS method is more sensitive than the kinetic method. It should be noted that this is the first AAS method for determination of the SLS. The kinetic method is comparable in accuracy and precision with the official method.
Acknowledgements
Authors thanks the Middle East pharmaceuticals and cosmetics laboratories, Palestine for supporting the salbutamol sulfate standard and drug formulation. As well thanking should be mentioned to Alaqsa University for offering all the required chemicals.References
- Martindale – the complete drug reference, ed. S. C. Sweetman, 36th edn, Pharmaceutical Press, London, 2009, p. 1125 Search PubMed.
- R. T. Sane, V. G. Nayak and V. B. Malkar, Talanta, 1985, 32, 31–33 CrossRef CAS.
- N. P. Sadler and H. Jacobs, Talanta, 1995, 42, 1385–1388 CrossRef CAS.
- R. S. Bakry, O. A. Razak, A. F. M. Walily and S. F. Belal, J. Pharm. Biomed. Anal., 1996, 14, 357–362 CrossRef CAS.
- D. Šatínský, R. Karlíček and A. Svoboda, Anal. Chim. Acta, 2002, 455, 103–109 CrossRef.
- G. G. Mohamed, S. M. Khalil, M. A. Zayed and M. A. El-Shall, J. Pharm. Biomed. Anal., 2002, 28, 1127–1133 CrossRef.
- I. Dol and M. Knochen, Talanta, 2004, 64, 1233–1236 CrossRef CAS PubMed.
- H. N. Dave, R. C. Mashru and A. R. Thakkar, Anal. Chim. Acta, 2007, 597, 113–120 CrossRef CAS PubMed.
- A. El-Gindy, S. Emara and H. Shaaban, J. Pharm. Biomed. Anal., 2007, 43, 973–982 CrossRef CAS PubMed.
- K. Basavaiah, B. C. Somashekar and V. Ramakrishna, Acta Pharm., 2007, 57, 87–98 CAS.
- P. Nagaraja, A. K. Shrestha, A. Shivakumar and A. K. Gowda, Acta Pharm., 2010, 60, 217–227 CrossRef CAS PubMed.
- S. S. Panda, B. V. Kumar and G. Mohanta, J. Pharm. Educ. Res., 2012, 3, 17–21 Search PubMed.
- H. N. Pandya, H. H. Berawala, D. M. Khatri and P. J. Mehta, J. Pharmacol. Toxicol. Methods, 2010, 1, 49–53 Search PubMed.
- Y. M. Issa, A. F. Shoukry and R. M. El-Nashar, J. Pharm. Biomed. Anal., 2001, 26, 379–386 CrossRef CAS.
- N. T. Abdel-Ghani, M. S. Rizk and R. M. El-Nashar, Anal. Lett., 2002, 35, 39–52 CrossRef CAS.
- D. Zhang, Y. Teng, K. Chen, S. Liu, C. Wei and B. Wang, Biomed. Chromatogr., 2012, 26, 1176–1182 CrossRef CAS PubMed.
- G. A. Jacobson and G. M. Peterson, J. Pharm. Biomed. Anal., 1994, 12, 825–832 CrossRef CAS.
- D. W. Boulton and J. P. Fawcett, J. Chromatogr. B: Biomed. Sci. Appl., 1995, 672, 103–109 CrossRef CAS.
- A. Halabi, C. Ferrayoli, M. Palacio and V. Dabbene, J. Pharm. Biomed. Anal., 2004, 34, 45–51 CrossRef CAS PubMed.
- S. V. Erram, C. B. Fanska and M. Asif, J. Pharm. Biomed. Anal., 2006, 40, 864–874 CrossRef CAS PubMed.
- G. B. Kasawar and M. Farooqui, J. Pharm. Biomed. Anal., 2010, 52, 19–29 CrossRef CAS PubMed.
- G. Wang, Y. Li, X. Li, X. Wang, Q. Guo, J. Wu, C. Xi and Z. Li, J. Chromatogr. Sci., 2011, 49, 276–280 CAS.
- A. S. Ghemud, B. Santhakumari, A. B. Pharne, M. M. Jadhay, K. S. Jain and M. Kulkarri, Int. J. Pharm. Pharm. Sci., 2012, 4, 249–253 CAS.
- K. Basavaiah and H. C. Prameela, Anal. Bioanal. Chem., 2003, 376, 879–883 CrossRef CAS PubMed.
- J. H. Espenson, Chemical Kinetics and reaction Mechanisms, McGraw-Hill, New York, 1981, p. 5 Search PubMed.
- G. Lopez-Cueto, M. Ostra and C. Ubide, Anal. Chim. Acta, 2001, 445, 117–126 CrossRef CAS.
- G. Lopez-Cueto, M. Ostra and C. Ubide, Anal. Bioanal. Chem., 2002, 372, 347–351 CrossRef CAS PubMed.
- G. Lopez-Cueto, M. Ostra and C. Ubide, Anal. Bioanal. Chem., 2002, 374, 915–922 CrossRef CAS PubMed.
- S. Wang, Q. Li, H. Bai and G. Liu, J. Chin. Chem. Soc., 2004, 51, 707–712 CrossRef CAS PubMed.
- J. Rose, Advanced Physico–Chemical Experiments, Pitman, London, 1964, p. 67 Search PubMed.
- British Pharmacopeia Vol. II, Her Majesty's Stationery Office, London, 1993, p. 584 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |