
Structure and properties of urea-formaldehyde resin/polyurethane blend prepared via in-situ polymerization
Junling Yuan,
Xiaowen Zhao* and
Lin Ye
State Key Laboratory of Polymer Materials Engineering, Polymer Research Institute of Sichuan University, Chengdu 610065, China. E-mail: zhaoxiaowenscu@126.com; Fax: +86-28-85402465; Tel: +86-28-85408802
First published on 1st June 2015
Abstract
Urea-formaldehyde resin/polyurethane (UF/PU) blends with improved toughness were prepared via in situ polymerization. It was found that both urea and PU participated in the addition and condensation reactions with formaldehyde, and thus a soft PU segment was chemically introduced into the UF system. Due to the relatively low reactivity of PU compared with urea, both the reaction activation energy (E) and the end temperature of the cure reaction increased with an increase in PU content. The blends showed a lower modulus, but a higher Tg compared with neat UF due to the improved degree of crosslinking in the blends. The toughness of UF can be obviously improved without a remarkable decrease in strength by introducing PU, resulting from the formation of a reaction-induced microphase-separated morphology and strong chemical interactions between the UF matrix and PU. The toughening mechanism of the blend corresponded to the energy-dissipation mechanism.
1. Introduction
Urea-formaldehyde (UF) resin is one of the most important types of so-called amino plastic resins, and is based on the manifold reaction between two monomers, urea and formaldehyde. Under different reaction conditions, a large variety of different condensed structures may be prepared. UF resin can be used as wood glue and surface coatings; it can be utilized to improve the wet strength of paper and the crease-resistance of textiles; powders from urea-formaldehyde resins can also be molded for use as containers, electrical fittings, bottle caps, lavatory seats and so on.1,2However, as a kind of thermosetting resin, UF is acknowledged to be very brittle and easily fractured during application. Therefore, despite the high strength and low-cost of UF, this drawback largely limits its usage. Many attempts have been made to improve the toughness of UF resins. One approach was to reduce the mole ratio of formaldehyde/urea, however, this lead to a decrease in the degree of crosslinking and thus, to an inferior performance of the resin, particularly regarding its mechanical strength and water resistance.3 Another approach to toughening UF resins is through resin modifiers, which have been widely used in the plastic industry. For example, PEG6000 was added into the UF matrix, and the impact strength of UF increased by 16% with 3% PEG content.4 Chen et al. prepared UF materials toughened with acrylate polymers, and their impact strength reached up to 2.5 kJ m−2.5 L. Yuan et al. synthesized a series of UF microcapsules containing epoxy resins which showed good storage stability, excellent solvent resistance and appropriate mechanical strength.6
Polyurethane elastomers (PUs), due to their unique property combination of high strength and high toughness, together with high damping, are widely used in many fields. PUs consist of thermodynamically incompatible hard and soft segments. The soft-segments largely control the toughness and low-temperature properties of PUs, while the hard-segments particularly provide the elastomer with high modulus, hardness, and tear strength.7,8 It is known that UF is prepared from the reaction of formaldehyde with the active hydrogen atom in the primary amine of urea. Encouragingly, there are plenty of secondary amines on the chains of PU, which make it possible to generate addition and condensation reactions between formaldehyde and PU, and thus the soft segments can be chemically introduced into the UF molecular chains and a good toughening effect would be achieved. To the best of our knowledge, few studies on UF/PU blends have been reported. There is just the work of Alan M. Wooler, which reported the fabrication of UF/PU foam with excellent heat- and flame-resistance by reacting an aqueous solution of an aminoplast precondensate with organic polyisocyanates in the presence of catalysts.9
In this work, PU was introduced to toughen UF resin via an in situ polymerization technique and the structures and properties of the UF/PU blends were studied. The effect of PU on the reaction kinetics of the UF resin polymerization were studied, and the toughening mechanism of PU on UF was explored.
2. Experimental
2.1 Materials
Urea was purchased from Bodi Chemical Ltd (Tianjin, China). Formaldehyde (37 wt% water solution) was purchased from Jinsan Chemical Reagent Co. Ltd (Chengdu, China). NaOH (sodium hydroxide) with analytical purity was purchased from Xilong (Chengdu, China). Ammonium chloride as the curing agent, was purchased from Bodi Chemical Ltd (Tianjin, China). The polyurethane (PU) used in this study was self-made in the laboratory.2.2 Synthesis and preparation
2.3 Measurements
The notched Charpy impact strengths of the neat UF and UF/PU blends were measured with a ZBC-4B impact testing machine from Xinsansi Co. (Shenzhen, China) according to ISO 179-1993.
3. Results and discussion
3.1 Synthesis and structure of the UF/PU blend
The evolution of the main chemical groups during the synthesis of the UF resin in the presence of PU was studied with in situ FTIR analysis.PU was mixed with formaldehyde to promote the reaction between them, and at this first stage, the time-dependent FTIR spectra in the 3100–2700 cm−1 and 1400–900 cm−1 regions were recorded and are shown in Fig. 1. The black curve was the last collected spectrum. It can be seen that the intensities of the peaks at 2795–2967 cm−1, assigned to symmetric and asymmetric stretching vibration of –CH2–, 1369 cm−1, assigned to the N–C stretching vibration, and 1105 cm−1, assigned to the C–O–C asymmetric bending vibration, all increased with increasing reaction time, indicating that reactions between formaldehyde and the –NH– groups of PU had occurred, and as shown in Fig. 2, the reaction may include two types of reactions: addition and condensation.11
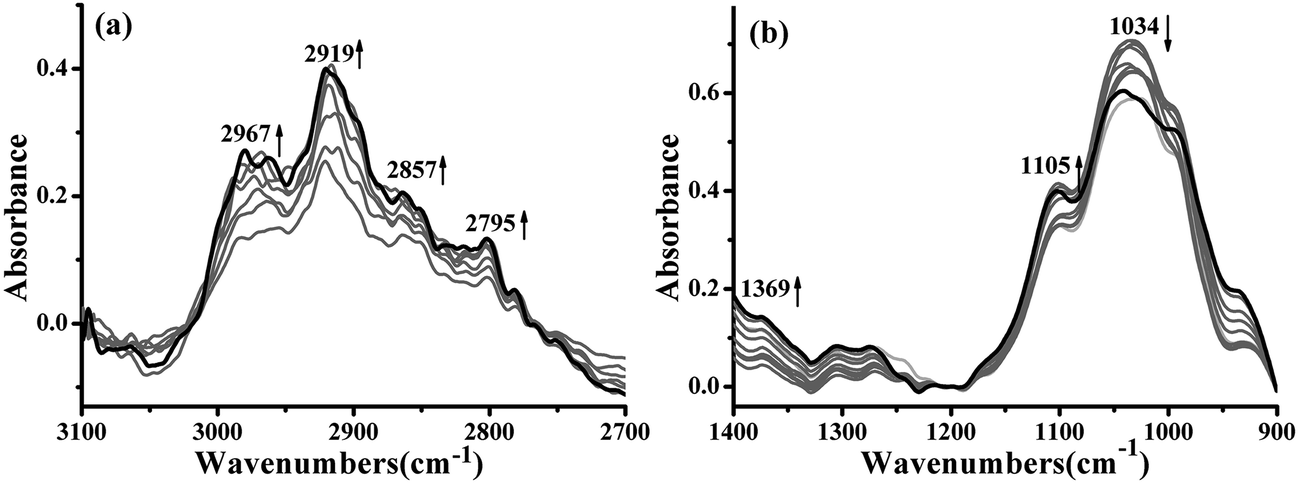 | ||
| Fig. 1 Time-dependent IR spectra in the (a) 3100–2700 cm−1 and (b) 1400–900 cm−1 regions of the UF/PU blend. The black curve was the last collected spectrum. Arrows indicate the changing trends of the corresponding peaks. | ||
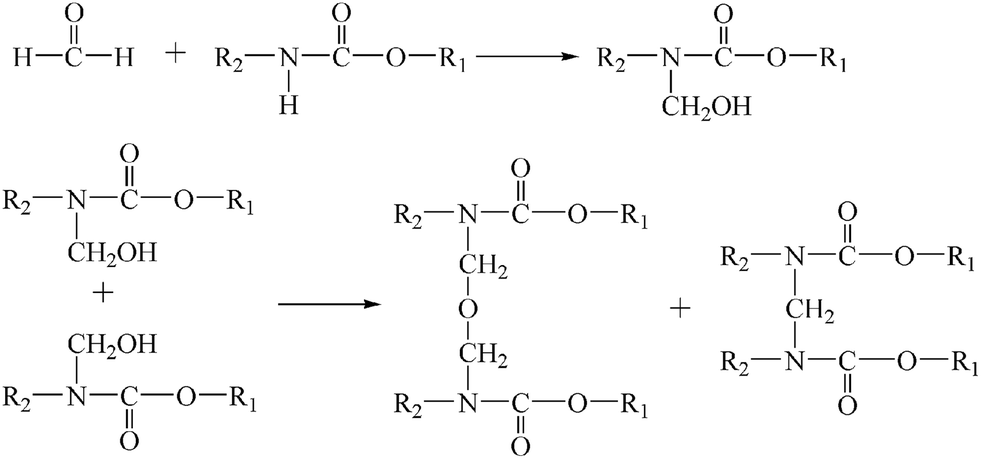 | ||
| Fig. 2 The reaction scheme for the reaction of PU with formaldehyde. | ||
After the reaction of PU with formaldehyde, urea was added into the PU–formaldehyde system, and the characteristic absorbances in the range of 1700–1100 cm−1 are shown in Fig. 3. The black curve was the last collected spectrum. It can be observed that the intensities of the peaks at 1544 cm−1, assigned to the –NH– bending vibration of the secondary amine group, 1354 cm−1, assigned to the N–C stretching vibration, and 1277 cm−1 assigned to the –CH2– bending vibration, increased with increasing reaction time, which indicated that an addition reaction had occurred between urea and formaldehyde during this stage. The intensity of the peak at 1134 cm−1, assigned to the C–O–C asymmetric bending vibration, also increased with reaction time, which indicated that a condensation reaction occurred simultaneously.12
 | ||
| Fig. 3 Time-dependent IR spectra in the 1700-1500 cm−1 region for the UF/PU blend after urea was added. The black curve was the last collected spectrum. Arrows indicate the changing trends of the corresponding peaks. | ||
For the final curing stage of the UF/PU system, the time-dependent FTIR spectra in the regions of 3800–3200 cm−1 and 1700–1000 cm−1 are shown in Fig. 4. The black curve was the last collected spectrum. For the pre-polymer, the broadening of the band at ∼3447 cm−1 could be assigned to the –OH stretching vibration of water and hydroxymethyl, or to the symmetric and asymmetric stretching vibrations of the –NH2 in the urea and the –NH– stretching vibration of the reacted urea or PU.13 With an increase in curing time, this peak shifted to a lower wavenumber and its intensity decreased, which resulted from the reaction of urea or PU with formaldehyde and the evaporation of water. The peak at 1620 cm−1, assigned to the –NH2 bending vibration, disappeared during this stage. The intensity of the peak at 1545 cm−1, assigned to the –NH– bending vibration, decreased, while the intensity of the peak at 1386 cm−1, assigned to the N–C stretching vibration, increased as the time increased, indicating the occurrence of condensation reactions between urea or PU and formaldehyde.
 | ||
| Fig. 4 Time-dependent IR spectra in the (a) 4000–3000 cm−1 and (b) 1700–1000 cm−1 regions for the UF/PU blend. The black curve was the last collected spectrum. Arrows indicate the changing trends of the corresponding peaks. | ||
3.2 Curing kinetics of the UF/PU blends
DSC analysis was used to further study the effect of PU on the curing process of UF. The DSC curves for all samples during curing at a heating rate of 5 °C/min are shown in Fig. 5, and the related data such as peak temperature (Tp) and half peak width (Δw) are listed in Table 1. In the case of the neat UF resin, the Tp was recorded as 120.3 °C, corresponding to the exothermic curing peak of the resin. While for the UF/PU blends, the Tps shifted to higher temperatures, which indicated that PU hindered the crosslinking reactions of the UF resins. Moreover, a new exothermic curing peak appeared at higher temperature for the UF/5 wt%-PU blend which may be assigned to the condensation reactions between PU and formaldehyde. Due to the relatively low reactivity of PU compared with urea, more energy was required for the crosslinking of the blends. With increasing PU content, the two peaks became closer and finally merged into a single one as shown for the UF/20 wt%-PU sample. | ||
| Fig. 5 The DSC curves for UF/PU blends with various PU content at a heating rate of 5 °C min−1. | ||
| Sample | Tp (°C) | Δw (°C) | E (kJ mol−1) | A (s−1) | n |
|---|---|---|---|---|---|
| Neat UF | 120.3 | 7.3 | 86.1 | 8.82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ×1010 ×1010 |
0.9293 |
| UF/5 wt%-PU | 125.2 | 11.1 | 91.5 | 3.57×1011 |
0.9340 |
| UF/10 wt%-PU | 128.5 | 12.6 | 101.6 | 5.82×1012 |
0.9373 |
| UF/15 wt%-PU | 136.5 | 28.6 | 139.3 | 3.07×1017 |
0.9438 |
| UF/20 wt%-PU | 149.3 | 35.3 | 1145.1 | 4.89×1017 |
0.9533 |
DSC analysis was further carried out at different heating rates, as shown in Fig. 6. The Tps shifted to higher temperatures as the heating rate increased for all samples. The kinetics of the curing reactions for the UF and UF/PU blends were studied using Kissinger’s method and Crane’s method:3
| ln(β/Tk2) = −E/RTk + ln(AR/E) | (1) |
| lnβ = −E/nRTk + C | (2) |
| Tk = Tp + 273.15 | (3) |
 | ||
| Fig. 6 The DSC curves at different heating rates for (a) the neat UF, (b) UF/5 wt%-PU, (c) UF/10 wt%-PU, (d) UF/15 wt%-PU and (e) UF/20 wt%-PU. | ||
In eqn (1), β is the heating rate; E is the activation energy; R is the gas constant with a value of 8.314 J K−1 mol−1 and A is the pre-exponential factor. In eqn (2), n is the reaction order and C is a constant.
The fitting curves for the Kissinger and Crane models were plotted in Fig. 7, and the kinetic parameters such as E, A and n could be calculated as shown in Table 1. The reaction orders n for neat UF and all of the blends were approximated to 1, indicating that the curing reaction of the UF resin was a one-step reaction. E increased with increasing PU content, suggesting that it was more difficult for the blend to cure.
 | ||
| Fig. 7 (a) Kissinger’s and (b) Crane’s plots for the neat UF and UF/PU blends with various PU content. | ||
The degree of curing for the neat UF and UF/PU blends can be calculated from the following equation.14
 | (4) |
As shown in Fig. 8, the DSC curves can be converted to the function XC(T) − T. All of the samples gave similar ‘‘S’’ shaped curves, and with an increase in curing temperature, XC(T) increased. Neat UF could cure at a relatively low temperature, and with increasing PU content, the end temperature of the cure reaction increased for the blends.
 | ||
| Fig. 8 The degree of curing for neat UF and the UF/PU blends with various PU content at a heating rate of 5 °C min−1. | ||
3.3 Dynamic mechanical properties of the UF/PU blends
The dynamic mechanical properties of neat UF and the UF/PU blends are shown in Fig. 9 and the characteristic parameters are listed in Table 2. The storage modulus of the blends decreased with increasing PU content, which suggested that the stiffness of the materials decreased. The tanδ curve for neat UF exhibited two relaxation peaks at about 24.97 °C and 130.51 °C corresponding to its β relaxation and α relaxation respectively. The α relaxation was aroused by chain segmental motion of the molecules corresponding to the glass transition temperature (Tg) and the β relaxation was aroused by the lateral group of molecules. In the case of the UF/PU blends, except for these two relaxations, a new peak appeared at about −60 °C due to the chain segmental motion of the soft segment of PU.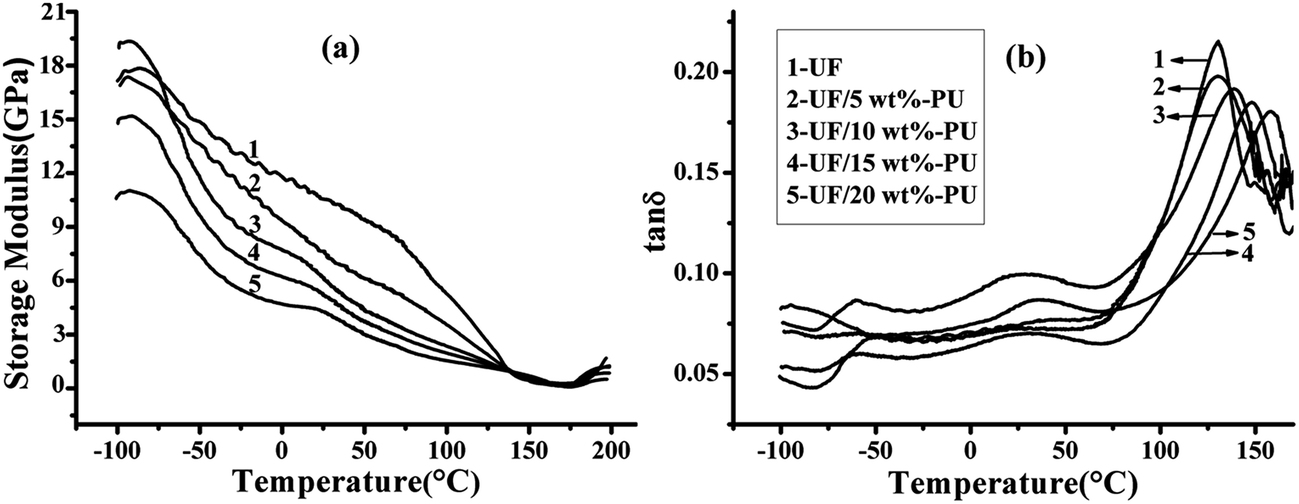 | ||
| Fig. 9 Dynamic mechanical properties of the UF/PU blends with various PU content. | ||
| Sample | Tg of PU (°C) | β relaxation temperature of UF (°C) | α relaxation temperature of UF (°C) | tanδmax |
|---|---|---|---|---|
| neat UF | — | 24.9 | 130.5 | 0.215 |
| UF/5 wt%-PU | −59.9 | 25.9 | 131.0 | 0.198 |
| UF/10 wt%-PU | −60.6 | 28.5 | 138.9 | 0.192 |
| UF/15 wt%-PU | −58.4 | 30.4 | 148.2 | 0.185 |
| UF/20 wt%-PU | −58.9 | 38.3 | 158.1 | 0.180 |
As shown in Table 2, with increasing PU content, the α and β relaxation temperatures for UF both increased, while tanδmax decreased gradually. For a typical polymer, Tg is inversely proportional to polymer chain mobility – low chain mobility results in a high Tg and also means that it is more difficult for the polymer to deform, and therefore a higher modulus is expected.15 However, experimental findings showed the opposite result in this study: the blends with high PU content showed a low modulus, but a high Tg. As mentioned, it can be deduced from the results of the in situ FTIR and curing kinetics for the UF/PU blends that abundant –NH– on the PU chains participated in the addition and curing reaction of UF, and the degree of crosslinking within the blend was thus improved. A schematic diagram of the UF/PU network is shown in Fig. 10.
 | ||
| Fig. 10 Crosslinking network of UF/PU blends. | ||
3.4 The toughening effect of PU on UF resin
The mechanical properties of the neat UF and UF/PU blends were determined by tensile and impact tests and the results are given in Fig. 11. It can be seen from Fig. 11(a) that although no obvious yield occurred for both the neat UF and UF/PU blended resins, the stress–strain curves of the samples varied with PU content. Young’s modulus decreased, while the strain at fracture increased with increasing PU content. | ||
| Fig. 11 The mechanical properties of the UF/PU blends with various PU content. | ||
It can be seen from Fig. 11(b and c) that the elongation at break and the notched impact strength of the blends increased obviously with increasing PU content, indicating that the UF resin was significantly toughened, and at the same time the tensile strength of the blends was still maintained at a high level.
SEM was used to characterize the fracture surface morphologies of neat UF and the UF/PU blends under two conditions: (i) impact fracture and (ii) cryogenic fracture. The images shown in Fig. 12 are the impact fracture surface morphologies of neat UF, UF/10 wt%-PU and UF/20 wt%-PU. It can be seen that the fracture surface of neat UF was relatively smooth and partly covered with striations, which is characteristic of a typical brittle fracture. With the addition of PU, the fracture surface changed from having a few small striations to many big striations and became rough. Many yield folds appeared, accompanied by a large number of major deformations from absorbing the impact energy, which is characteristic of a relatively tough fracture.16 The most obvious deformation occurred at 20 wt%-PU content, and a remarkably enhanced toughness was presented.
 | ||
| Fig. 12 The SEM images of the impact fractured surfaces of (a) neat UF, (b) UF/10 wt%-PU and (c) UF/20 wt%-PU (magnification: ×2000). | ||
The cryogenic fracture surfaces for neat UF and the UF/PU blends are shown in Fig. 13. For the neat UF resin, a featureless morphology was exhibited, while for the UF/20 wt%-PU, a microphase-separated morphology was observed. The dark continuous regions were attributed to the UF matrix, whereas the light regions were the PU domains. The spherical PU particles and some coagulated worm-like PU domains with a size of about 50 nm were homogeneously dispersed in the matrix.
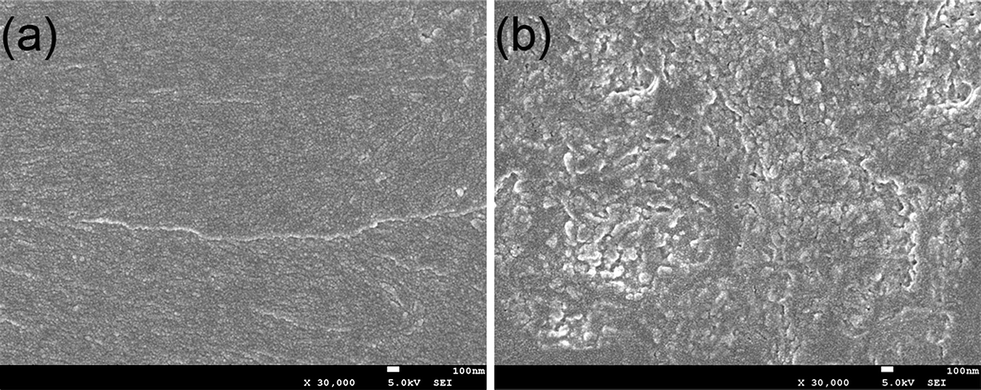 | ||
| Fig. 13 The SEM images of the cryogenic fracture surfaces of (a) neat UF and (b) UF/20 wt%-PU (magnification: ×30000). | ||
The formation of this kind of microphase-separated morphology in UF thermosets containing block copolymers could have occurred by the following mechanism: before curing, the mixtures composed of formaldehyde, PU and urea, were transparent, suggesting that the mixtures were homogeneous and that no macroscopic phase separation had occurred on a scale larger than the wavelength of visible light. With the reaction proceeding, –NH– on the PU chains participated in the addition, condensation and curing reactions of UF, and thus these parts of the PU subchains could be interpenetrated into the cross-linked UF network at the segmental level. However, the soft-segments of the PU chains were high molecular weight polyether or polyester macroglycols, which tended not to be involved in any reaction during the synthesis of UF. With the reactions going on, the Gibbs free energy of mixing increased due to the decrease in entropy. And when the free energy increased to a certain degree, the soft segment of PU could be separated out, whereas the NH subchains remained in the cross-linked UF networks, resulting in the microphase separation.
Therefore, it can be concluded that the improvement in fracture toughness of the UF/PU blends mainly resulted from the formation of a reaction-induced microphase-separated morphology as well as from the strong chemical interactions between the UF matrix and PU.17 The PU domains, with high toughness, could serve as stress concentrators to trigger and stabilize massive crazes in the UF matrix. At the same time, the –NH– on the PU chains that were involved in the reactions with UF, served as a bridge between the UF and PU phases, which was beneficial for the ability of PU to transfer and disperse the stress induced when the UF matrix was subjected to an external force, thus corresponding to the energy-dissipation mechanism.18
4. Conclusions
In order to improve the toughness of UF, the UF/PU blends were prepared via in situ polymerization. It was verified by in situ FTIR analysis that the NH groups within the PU molecules participated in two types of reactions (addition and condensation) with formaldehyde. The curing reactions of the UF resin, both in the presence and absence of PU, were one-step reactions. Due to the relatively low reactivity of PU compared with urea, both the reaction activation energy (E) and the end temperature of the cure reaction increased with the increase in PU content. The blends showed a lower modulus, but a higher Tg compared with neat UF due to the improved degree of crosslinking within the blends. The elongation at break and the notched impact strength of the blends increased with increasing PU content, while the tensile strength was still maintained at a relatively high level. By the addition of PU, the fracture surface changed from having a few small striations to many big striations and became rough, suggesting that a remarkably enhanced toughness was presented. A homogeneous microphase-separated morphology could be observed for the blends, which was believed to be critical for improving the toughness of the blend. The toughening mechanism of the blend corresponded to the energy-dissipation mechanism.References
- G. I. Gavrilović, O. Nešković and M. M. Điporović, Molar-mass distribution of urea-formaldehyde resins of different degrees of polymerization by MALDI-TOF mass spectrometry, J. Serb. Chem. Soc., 2010, 75(5), 689–701 CrossRef.
- A. S. Singha and V. K. Thakur, Synthesis and characterization of short Grewia optiva fiber-based polymer composites, Polym. Compos., 2010, 31(3), 459–470 CAS.
- E. Roumeli and E. Papadopoulou, Synthesis, characterization and thermal analysis of urea–formaldehyde/nano SiO2 resins, Thermochim. Acta, 2012, 527, 33–39 CrossRef CAS PubMed.
- C. G. Han and S. H. Li, Effects of different plasticizers on performance of urea formaldehyde molding plastics, Mod. Chem. Ind., 2011, 31(2), 61–65 CAS.
- R. Chen, et al., CN Pat., 201310745020.7, 2013.
- L. Yuan, A. J. Gu and G. Z. Liang, Preparation and properties of poly (urea–formaldehyde) microcapsules filled with epoxy resins, Mater. Chem. Phys., 2008, 110(2), 417–425 CrossRef CAS PubMed.
- J. Guan, Y. Song and Y. Lin, Progress in study of non-isocyanate polyurethane, Ind. Eng. Chem. Res., 2011, 50(11), 6517–6527 CrossRef CAS.
- J. Wang and L. Ye, Structure and properties of polyvinyl alcohol/polyurethane blends, Composites, Part B, 2015, 69, 389–396 CrossRef CAS PubMed.
- A. M. Wooler, U.S. Pat., 4 435 526, 1984.
- B. D. Park and H. W. Jeong, Hydrolytic stability and crystallinity of cured urea-formaldehyde resin adhesives with different formaldehyde/urea mole ratios, Int. J. Adhes. Adhes., 2011, 31(6), 524–529 CrossRef CAS PubMed.
- K. Siimer, T. Kaljuvee and P. Christjanson, Thermal behaviour of urea-formaldehyde resins during curing, J. Therm. Anal. Calorim., 2003, 72(2), 607–617 CrossRef CAS.
- B. Likozar, R. C. Korošec and I. Poljanšek, Curing kinetics study of melamine-urea-formaldehyde resin, J. Therm. Anal. Calorim., 2012, 109(3), 1413–1422 CrossRef CAS PubMed.
- T. Zorba, E. Papadopoulou and A. Hatjiissaak, Urea-formaldehyde resins characterized by thermal analysis and FTIR method, J. Therm. Anal. Calorim., 2008, 92(1), 29–33 CrossRef CAS.
- S. Xu, X. W. Zhao and L. Ye, Mechanical and crystalline properties of monomer casting Nylon-6/SiO2 composites prepared via in situ polymerization, Polym. Eng. Sci., 2013, 53(9), 1809–1822 CAS.
- J. Zheng, R. Ozisik and R. W. Siegel, Disruption of self-assembly and altered mechanical behavior in polyurethane/zinc oxide nanocomposites, Polymer, 2005, 46(24), 10873–10882 CrossRef CAS PubMed.
- S. Xu, X. W. Zhao and L. Ye, Preparation and Properties of Monomer Casting Nylon-6/PEO Blend Prepared via In-situ Polymerization, Polym. Eng. Sci., 2013, 53(9), 1809–1822 CAS.
- F. L. Meng, S. X. Zheng and H. Q. Li, Formation of ordered nanostructures in epoxy thermosets: a mechanism of reaction-induced microphase separation, Macromolecules, 2006, 39(15), 5072–5080 CrossRef CAS.
- W. Gong, K. Zeng and L. Wang, Poly(hydroxyether of bisphenol A)-block-polydimethylsiloxane alternating block copolymer and its nanostructured blends with epoxy resin, Polymer, 2008, 49(15), 3318–3326 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |