
Assembly of Zn-metal organic frameworks based on a N-rich ligand: selective sorption for CO2 and luminescence sensing of nitro explosives†
Abstract
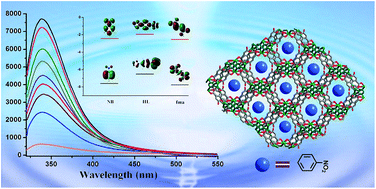
Two novel Zn(II) coordination polymer compounds, namely, [Zn3(HL)2(fma)2]·DMA·H2O (1) and [Zn2(μ3-OH)(HL)(Br-bdc)]·0.5DMA·CH3OH·H2O (2) have been successfully obtained with 1-(5-tetrazolyl)-4-(1-imidazolyl) benzene ligand and dicarboxylic acids (fma = fumaric acid; Br-bdc = 2-bromoterephthalic acid). Compound 1 exhibits a 3D framework with a square aperture diameter for the channel is 8.8 × 8.8 Å2, and the framework can be simplified as a (3,4)-connected tfj network with the point symbol of {4.82}4{42.84}2{84.122} topology. In compound 2, the HL ligand forms a 2D sheet of Zn(HL) by linking Zn ions along the c axis, which are further connected by double Br-bdc pillars resulting in unique bi-pillared-layer type 3D frameworks. The result, 1a, exhibits a certain degree of CO2 uptake and selective CO2/N2 adsorption capacity. Furthermore, luminescent properties of 1a well dispersed in different solvents have also been investigated systematically, which demonstrate distinct solvent-dependent luminescent spectra with emission intensities significantly quenched toward acetone and nitrobenzene.
 Please wait while we load your content...
Please wait while we load your content...

