
Monodisperse platinum nanoparticles supported on highly ordered mesoporous silicon nitride nanoblocks: superior catalytic activity for hydrogen generation from sodium borohydride†
Abstract
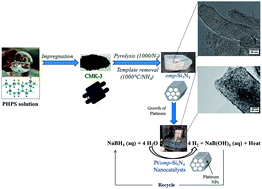
Late transition metal have attracted considerable interest for catalytic applications. Their immobilization over supports with tailored porosity is advantageous for nanosizing metal particles and avoiding their agglomeration which is known to bring a serious issue to the catalytic performance. Herein, ordered mesoporous silicon nitride (Si3N4) nanoblocks with hexagonal symmetry of the pores, high specific surface areas (772.4 m2 g−1) and pore volume (1.19 cm3 g−1) are synthesized by nanocasting using perhydropolysilazane as precursor. Then, Si3N4 nanoblocks are used as supports to synthesize platinum nanoparticles (Pt NPs) by precursor wet impregnation. Detailed characterizations by TEM show that monodispersed spherical Pt NPs with a 6.77 nm diameter are successfully loaded over nanoblocks to generate nanocatalysts. The latter are subsequently used for the catalytic hydrolysis of sodium borohydride (NaBH4). A hydrogen generation rate of 13.54 L min−1 gPt−1 is measured. It is notably higher than the catalytic hydrolysis using Pt/CMK-3 nanocatalysts (2.58 L min−1 gPt−1) most probably due to the textural properties of the Si3N4 supports associated with the intrinsic properties of Si3N4. This leads to an attractive nanocatalyst in pursuit of practical implementation of B-/N-based chemical hydrides as a hydrogen source for fuel cell application.
 Please wait while we load your content...
Please wait while we load your content...

