
Hydrothermal synthesis and enhanced photocatalytic activity of ternary Fe2O3/ZnFe2O4/ZnO nanocomposite through cascade electron transfer†
Abstract
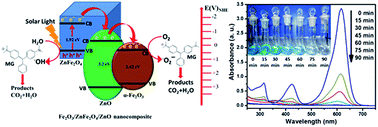
This work represents the synthesis and photocatalytic activity of α-Fe2O3 nanoparticles and mixed oxide nanocomposites such as Fe2O3/ZnFe2O4, Fe2O3/ZnFe2O4/ZnO and ZnFe2O4/ZnO. The above mentioned nanomaterials were synthesized by hydrothermal method by varying the molar ratio of iron and zinc salts followed by calcination at 500 °C. The crystalline phase, surface morphology and size of the prepared nanomaterials were characterized by XRD, FESEM & TEM analysis. FESEM and TEM images indicate the formation of irregular shape for α-Fe2O3 and cubical shape for nanocomposite systems having diameter in the range of 80–100 nm. The prepared nanomaterials were used for photocatalytic degradation of malachite green in aqueous media under solar light irradiation. The nanocomposites exhibit enhanced photocatalytic activity as compared to α-Fe2O3 nanoparticles. However, Fe2O3/ZnFe2O4/ZnO (Fe : Zn = 70 : 30) nanocomposite exhibits highest photocatalytic activity. This result might be due cascade electron transfer mechanism in the ternary phase.
 Please wait while we load your content...
Please wait while we load your content...

