
Biophysical, biopharmaceutical and toxicological significance of biomedical nanoparticles
Sangeetha Aula
ab,
Samyuktha Lakkireddy
ab,
Kaiser Jamil
a,
Atya Kapley
ac,
A. V. N. Swamy
d and
Harivardhan Reddy Lakkireddy
*e
aCentre for Biotechnology and Bioinformatics, Jawaharlal Nehru Institute of Advanced Studies (JNIAS), Secunderabad, Telangana, India
bDepartment of Biotechnology, Jawaharlal Nehru Technological University Anantapur (JNTUA), Anantapuramu, Andhra Pradesh, India
cEnvironmental Genomics Division, Council of Scientific and Industrial Research-National Environmental Engineering Research Institute (CSIR-NEERI), Nagpur, Maharashtra, India
dDepartment of Chemical Engineering, Jawaharlal Nehru Technological University Anantapur (JNTUA), Anantapuramu, Andhra Pradesh, India
eDrug Delivery Technologies and Innovation, Pharmaceutical Sciences, Sanofi Research and Development, 13 Quai Jules Guesde, 94403 Vitry-sur-Seine, France. E-mail: harivardhan-reddy.lakkireddy@sanofi.com
First published on 8th May 2015
Abstract
Nanotechnology has undoubtedly brought innovation to the biomedical field, which is apparent from the advances including those in drug delivery, treatment of pathologies, imaging of disease sites, etc. The rationale behind the use of nanoparticle-based products for biomedical applications is to benefit from their unique physicochemical characteristics, mainly, size, surface area and surface functionality, to address these particles and the encapsulated payload, if any, to the desired sites in the biological system. To design appropriate nanoparticle products for biomedical applications aimed for human and/or animal use, understanding of the interplay between the physicochemistry of nanoparticles and the biophysical properties is crucial because it is the interaction of the nanoparticles at the biological interface which regulates the nanoparticles pharmacokinetics, biodistribution and safety. Also, the assessment of the potential of nanoparticles to induce undesired effects at the systemic level, organ level, cellular and sub-cellular levels is crucial for anticipating the potential risks associated with the use of nanoparticles from a safety standpoint. This review is aimed at summarizing the nanoparticle candidates for biomedical applications, and reviewing, based on the relevant literature data, the inter-relationship between nanoparticles' physicochemistry and biophysical properties in conditioning the nano–bio interactions and inturn regulating the nanoparticles pharmacokinetics, biodistribution and toxicological properties. Besides, the importance of designing relevant physiologically-based modeling approaches for the simulation and prediction of the performance and safety of new nanomaterials based on their properties has been also discussed. An important portion of the review focusses also on the description of the methodologies for a detailed assessment of the toxicological properties of the nanoparticles.
 Sangeetha Aula | Sangeetha Aula is a Ph.D. scholar at the School of Life Sciences (SLS), Jawaharlal Nehru Institute of Advanced Studies (JNIAS), Secunderabad, affiliated to JNTUA, Andhra Pradesh, India. She has a Masters degree in Biochemistry and is currently pursuing Ph.D. Her research interests include assessment of the interaction of nanomaterials, organic and inorganic, with biological environment in vitro and in vivo. |
 Samyuktha Lakkireddy | Samyuktha Lakkireddy is a research scholar in the School of Life Sciences (SLS), Jawaharlal Nehru Institute of Advanced Studies (JNIAS), Secunderabad, affiliated to JNTUA, Andhra Pradesh, India. She has a Masters degree in Biochemistry, and is currently pursuing Ph.D. Her research interests include cancer biology, molecular genetics. |
 Kaiser Jamil | Dr Kaiser Jamil, Dean, School of Life Sciences, JNIAS had the opportunity to work in the country's foremost CSIR labs (CCMB & IICT), which gave her an exposure to professional scientific environment. She had the privilege to interact with some distinguished national and international scientists and Nobel Laureates. Presently she is dedicatedly working in the thrust areas like cancer biology and Nanotechnology and has published more than 200 papers in journals of repute. She presented her work as plenary and invited lectures at National & International conferences in different countries, and is a recipient of several awards and honors. |
 Atya Kapley | Dr (Mrs) Atya Kapley, Ph.D. from University of Hyderabad, Hyderabad, Principal Scientist at the National Environmental Engineering Research Institute, Nagpur, works in the field of environmental genomics. She uses a multi-disciplinary approach, combining conventional microbiology tools with bioinformatics and molecular tools to address rising levels of environmental contamination. She has worked on the bioremediation of pesticide contaminated soil, and uses metagenomics approach to study microbial communities in activated biomass of wastewater treatment plants with the aim to understand and improve biological treatment capacity. |
 A. V. N. Swamy | Dr A. V. N. Swamy obtained his Ph.D. from IIT Bombay. He worked as a professor in the department of chemical engineering in Jawaharlal Nehru Technological University Anantapur (JNTUA), Anantapuramu, Andhra Pradesh, India. He worked for about 20 years in various process industries, prior to joining JNTUA. His research areas include optimizing production of amino acids through fermentation, application of biotechnology in the treatment of industrial waste water. He has published more than 20 papers in reputed journals. He authored a book on “Fundamentals of Biochemical engineering”, BS publications, Hyderabad, 2007. He had successfully completed research projects funded by UGC, AICTE. |
 Harivardhan Reddy Lakkireddy | Harivardhan Reddy Lakkireddy is the Head of Drug Delivery Technologies & Innovation-Nanotechnologies, at Sanofi Research & Development, France. He has a Ph.D. in nanomedicine, and postdoc from faculty of Pharmacy, University of Paris-Sud XI, France. He has 55 peer reviewed publications and 5 patents to his credit, and co-edited a book. He is a member of various nanotechnology and drug delivery societies. His interests are nanomedicines, drug delivery, biopharmaceutics and pharmaceutical development. |
1. Introduction
Significant advances in nanotechnology in the field of biomedicine have resulted in a variety of smart innovations, especially in the areas of disease therapy and imaging, many of which have been transformed successfully into clinically applicable products. Biomedical nanotechnology has been an inter-disciplinary field orchestrated by physics, biology, chemistry, engineering, pharmacy, medicine etc., to translate the idea from the brain and bench to the bedside.In disease therapy, the nanotechnology has an important contribution to the transformation of the way the drugs challenging from physicochemical and biopharmaceutical standpoints have been delivered, and also the treatment of pathologies using nanoparticles per se.1–3 For imaging, nanotechnology has been successfully employed in clinical situations in imaging of various pathologies, with improved performance.4–6 Such successful milestones in biomedical nanotechnology have led to the continued motivation of researchers to design and explore a variety of new materials (e.g. polymers, lipids, inorganic materials, and hybrid composites), architectures, and functionalities for enhanced performance, responsive to different stimuli, compatible with biological milieu, etc. for addressing the evolving biomedical challenges, as evident from the intensive literature and intellectual property published each year in this area. While the performance of such new nanomaterials is important from their effectiveness standpoint, understanding of these nanomaterials with increasing complexity, from structural, physicochemical, biophysical, biopharmaceutical and toxicological standpoints, and their inter-play, is crucial to be able to develop performing and safer nanoparticle products and to ensure the product with reproducible quality attributes at each stage of development for intended application. Thus, in this review, we summarized the importance of the above aspects in the nano–bio interaction space, including the key considerations and assays for a detailed assessment of the toxicological properties of the nanoparticles, both in in vitro and in in vivo settings.
2. Nanoparticles for biomedical applications
Biomedical nanotechnology, which involves the design and development of nanoparticles for various biomedical applications, has encountered a significant pace of development in the recent years as evident from a variety of nanotechnology-based products commercialized and in clinical and preclinical testing for biomedical purposes. The nanoparticles for biomedical applications could be classified as indicated in the Fig. 1. | ||
| Fig. 1 Classification of nanoparticles for biomedical applications. | ||
The principal applications for which the nanoparticles have been widely employed include the delivery of medicinal substances (prophylactic or therapeutic),7,8 for disease therapy by local induction of stimuli in the target tissues,9,10 for imaging,6,11 for simultaneous therapy and imaging (termed as 'theranostic'),12,13 for tissue engineering,14 for detoxification,15,16 etc.
The main focus of nanoparticles application in the recent years has been on drug delivery (e.g. therapeutic agents, prophylactic agents, antigens, genes). This area has advanced significantly with the introduction of a variety of approaches to address specific challenges, such as, improvement of dissolution of poorly soluble drug molecules17,18 enable the administration of poorly soluble drugs,19,20 alter the drug pharmacokinetics to achieve desired therapeutic efficacy and safety,21,22 site-specific delivery of the drugs to specific organs/tissues of interest,23–25 and enable the intracellular delivery of difficult-to-deliver drugs,26,27 such as, nucleic acids, proteins and peptides. In case of vaccines, the nanoparticles have been employed as carriers for delivering antigens and/or immune adjuvants for enhancing their presentation to the antigen-presenting cells thereby contributing to the efficient induction of the immune responses.28,29 In drug discovery and development, the nanoparticles have been also considered, (i) as screening tools at the drug discovery stage to identify molecules of significant therapeutic activity and those suitable for further development to human, (ii) as translational tools for understanding the biological mechanism of a disease or for validation of disease targets, and (iii) to develop added-value therapeutic versions of the patent expired drugs for life cycle management.1,30 Thus, it is not exaggerating to mention that the nanoparticles are becoming integrated tools in discovery and development in pharmaceutical and biotechnology areas.
Additionally, the nanoparticles have been well demonstrated for use in diagnostic imaging, as magnetic resonance contrasts or as fluorescence agents, either per se or as carriers, to track the nanoparticle biodistribution in the body or for detection of pathologies in the body. This is evident from a variety of products approved for commercialization for human application, based mainly on iron oxide,6 such as, ferumoxsil, ferumoxide, ferrixan, ferristene, and from those products in early stages of proof of concept for near-infrared fluorescence imaging, magnetic resonance imaging, positron emission tomography, ultrasound imaging, etc.31,32 Recently, considerable emphasis has been made about designing of the inorganic nanoparticles with renal clearance properties for use as contrast agents.33–35
The concept of nanotheranostics,12,36,37 wherein the nanoparticle-based systems with combined capability of drug delivery and imaging, i.e., addressing the drug to the target tissue/cell and simultaneously allowing the monitoring of response to the treatment, is emerging at a good pace in the context of personalized medicine.
The details of nanotechnology-based products commercialized for drug delivery and imaging have been provided in the Table 1.
| Product | Technology | Route | Indication |
|---|---|---|---|
| Improve drug bioavailability/enable drug administration | |||
| Rapamune | Nanocrystalline suspension of rapamycin | Oral | Immunosuppressive |
| Tricor | Nanocrystalline suspension of fenofibrate | Oral | Hypercholesterolemia |
| Triglide | Nanocrystalline suspension of fenofibrate | Oral | Hypercholesterolemia |
| Emend | Nanocrystalline suspension of aprepitant | Oral | Nausea and emesis |
| Megace ES | Nanocrystalline suspension of megestrol | Oral | Anorexia |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Alter drug pharmacokinetics/achieve therapeutic benefit | |||
| Marqibo | Sphingomyelin/cholesterol liposome (non-PEGylated) containing vincristine sulfate | Intravenous | Cancer |
| Abraxane | Albumin nanoparticle loaded with paclitaxel | Intravenous | Cancer |
| Invega Sustenna | Drug nanocrystalline suspension | Intramuscular | Schizophrenia |
| DepoCyt | Liposome containing cytarabine | Intravenous | Cancer |
| Ambisome | Liposome (non-PEGylated) containing amphotericin B | Intravenous | Visceral leishmaniasis, fungal infections |
| Doxil/Caelyx | Liposome (PEGylated) containing doxorubicin HCl | Intravenous | Cancer |
| DaunoXome | Liposome (non-PEGylated) containing daunorubicin | Intravenous | Cancer |
| Myocet | Liposome (non-PEGylated) containing doxorubicin citrate | Intravenous | Cancer |
| Diprivan | Emulsion containing propofol | Intravenous | Anesthetic |
|
|||
| Delivery of antigens/vaccines | |||
| Fluad | Squalene-based oil-in-water nano-emulsion formulation containing influenza/pandemic flu virus antigen | Intramuscular | Influenza |
| Pandemrix | AS03 adjuvant (oil-in-water emulsion made of α-tocopherol, squalene and polysorbate 80) loaded with H1N1 influenza antigen | Intramuscular | H1N1 influenza pandemic (flu) |
| Fendrix | AS04 adjuvant (dispersion of monophosphoryl lipid A and aluminium phosphate) loaded with hepatitis B surface antigen | Intramuscular | Hepatitis B viral infection |
| Epaxal | Reconstituted viral membrane vesicles containing the viral proteins and lipids loaded with inactivated hepatitis A virus | Intramuscular | Hepatitis A virus infection |
| Inflexal® V | Reconstituted viral membrane vesicles containing the viral proteins and lipids loaded with influenza (subunit) | Intramuscular | Influenza |
| Cervarix | AS04 adjuvant (dispersion of monophosphoryl lipid A and aluminium phosphate) loaded with human papilloma virus antigens (types 16 and 18) | Intramuscular | Cervical cancer caused by human papilloma virus |
| Gardasil | Virus-like particles containing human papilloma virus antigens (types 6, 11, 16, and 18) | Intramuscular | Cervical cancer caused by human papilloma virus |
|
|||
| Imaging | |||
| Ferumoxsil/Lumirem | Iron oxide nanoparticle coated with dextran | Oral | Gastrointestinal imaging |
| Ferristene/Abdoscan | Iron oxide nanoparticle coated with sulfonated styrene–divinylbenzene copolymer | Oral | Gastrointestinal imaging |
| Ferumoxide/Endorem | Iron oxide nanoparticle coated with dextran | Intravenous | Liver imaging |
| Ferucarbotran/Resovist | Iron oxide nanoparticle coated with dextran | Intravenous | Liver imaging |
3. When nanoparticles encounter blood circulation
Following the exposure of the body to nanoparticles, be it through skin or by inhalation or by oral uptake or by injection, after the first point of contact, i.e., skin membrane after skin exposure, the nasal epithelium, lung epithelium and the pulmonary cells after inhalation, the stomach and intestinal epithelia after oral uptake, and the blood following injection, the nanoparticles either distribute to the local tissues or are potentially transported to the systemic circulation and subsequently to the various tissues in the body. When nanoparticles enter into contact with blood, various interactions could be described within the nano–bio interaction space, as below.3.1. Interaction with blood/plasma proteins
The fundamentally important principle in the context of nanoparticles is the size and surface area relationship. The particle surface area and the size are inversely related, thus the surface area increases with decreasing particle size. Surface property of nanoparticles is another important factor influencing the nano–bio interactions. When the nanoparticles enter into contact with the blood circulation, the blood components, such as, plasma proteins and other biomolecules compete for binding onto the nanoparticle surface resulting in the formation of the nanoparticle–protein complexes with protein corona on the nanoparticles surface. Thus, the surface property of the nanoparticles before coming into contact with blood may turn out to be different after their contact with blood. Depending on the affinity of blood/plasma proteins to the nanoparticle surface, the proteins may adsorb and desorb in a dynamic fashion leading to a corona, and at a given point of time, the protein corona may be either soft (containing reversibly binding proteins with faster exchange rate) or hard (containing irreversibly binding proteins with slower exchange rate).38,39 The kinetics of nanoparticle–protein association and dissociation process may have important roles in determining the particle's interactions with biological surfaces and the receptors and thus the nanoparticles overall fate.40 Furthermore, the proteins adsorbed onto the nanoparticle surface may undergo conformational change and consequently may exhibit functional change.41 The nanoparticles often curved and exhibiting high surface area may stimulate the proteins to alter structurally to enable occupying the surface. For instance, binding to nanoparticles resulted in conformational change of various proteins, such as, albumin,42 tubulin,43 transferrin,44 etc. Such undesired conformational changes in the protein structure may have important implications in terms of protein–protein interactions, cellular signaling and the related mechanisms. Thus, it is not surprising that the nanoparticles distributed to a tissue interact with the cells not with its original size and surface characteristics but with the newly acquired protein-corona induced size and surface characteristics. It means that the nanoparticles interaction with the cells within a tissue is probably more mediated by the nature and conformation of the surface bound proteins, and so as for the cellular internalization of the nanoparticles.45 So, the key question here is what drives the nanoparticle–protein interactions. It is becoming clearer that the nanoparticles' physicochemical characteristics prior to contact with the biological milieu influence the biophysical properties. Mainly, the nanoparticles' size and surface charge are the important decisive factors in differential protein adsorption and protein corona formation on the nanoparticles surface.46,47 The plasma protein binding onto the nanoparticles of size as small as 80 nm diameter (6% protein bound) was shown to be lower compared to that of the nanoparticles of 240 nm diameter (34% protein bound).48 In vivo, the intravenously injected nanoparticles of diameters ranging between 80–150 nm underwent rapid systemic clearance with a plasma half-life of 8–30 min as compared to the small sized nanoparticles of 20–40 nm which exhibited a plasma half-life of 25–30 h.49,50On the other hand, the neutral charged nanoparticles experienced less protein adsorption compared to that of the negative and positive charged nanoparticles.51 The nanoparticles size was also shown to influence the affinity of proteins to the particle surface and also the changes in protein structural conformation and function.52,53 Moreover, the protein adsorption onto the nanoparticles was also influenced by the nanoparticles shape prior to contact with the proteins.54
The biophysical properties of such nanoparticle–protein complexes in vivo influence the biodistribution of nanoparticles.55,56 Adsorption of opsonins such as complement, fibrinogen, immunoglubulinG (IgG), etc. is believed to promote macrophage uptake and subsequent phagocytosis resulting in the nanoparticles clearance from the systemic circulation,57,58 whereas, binding of dysopsonins such as human serum albumin, apolipoproteins etc. regulate the nanoparticles distribution and accumulation in specific tissues.59 Adsorption of apolipoproteins on the intravenously injected nanoparticles surface stimulate the interaction of nanoparticles with low density lipoprotein receptors, thus either promoting their transport across the blood–brain barrier60 or their accumulation in the liver parenchyma,61 tissues which have abundance of lipoprotein receptors. This suggests the importance of understanding the nano–bio interactions, and making the link between the physicochemical characteristics of nanoparticles and their biophysical properties, to be able to anticipate the nanoparticles performance and safety.
3.2. Interaction with immune system components
The nanoparticles encountering the blood stream also experience interaction with the components of the immune system, potentially resulting in the consequences, such as, the induction of complement activation, coagulation, and inflammatory response. Complement is one of the important components of the immune system acting as a ‘watch dog’ against the invading pathogens. Thus, the complement system acts as a first line of defense in the innate immunity and also play an important role in the induction and regulation of the adaptive B-cell and T-cell immune responses. Complement is a complex network of over 30 plasma and membrane proteins organized into a hierarchy of proteolytic cascades starting from the recognition of invading pathogens and the consequent steps of the immune activation.62 Whatever the pathways suggested for the activation of complement, they all result in the production of a major complement fragment C3b. C3b possess the ability to induce opsonization and also results in the complement effectors, by the action of C3 convertases to result in the formation of membrane attack complex C5b-9, and by the action of C5 convertases to result in anaphylotoxins C3a, C4a and C5a which mediate inflammation. While complement function is crucial for the body's immunity, the uncontrolled/excess activation of complement may result in anaphylactic reactions and even to death.63 Nanoparticles, due to their small size, extensive surface area, composition and functionality, possess great potential to interact with complement stimulating molecules and subsequently induce complement activation.64–66 Moreover, nanoparticles possessing different surface charge exhibited different abilities to induce complement activation. For instance, the positively charged particles induced higher levels of complement activation compared to negatively charged and neutral charged particles.67Moreover, the complement effectors formed in response to complement activation are shown to directly enhance blood coagulation. Such effect is supported by the inflammatory mediators leading to the thrombogenicity of blood. For instance, the anaphylatoxin C3a activates platelets thereby, enhancing their aggregation and adhesion, while the anaphylotoxin C5a enhances blood thrombogenicity.68 Complement and coagulation pathways activate each other, for instance, the thrombin formed during the coagulation pathway and the platelets are suggested to catalyse the amplification of complement. Complement was also reported to inhibit anticoagulant factors. In addition, the complement and coagulation are suggested to be the partners in inducing the inflammatory response.68
The anaphylatoxins C3a and C5a formed during the complement pathway have been suggested also to contribute to the regulation of the inflammatory cytokine response and influence the production and secretion of tumor necrosis factor-α and interleukin-6.69,70
Thus, the nanoparticles-mediated complement activation may be the cause of concern, as discussed above, due to the consequential undesired effects, such as, coagulation and mounting of inflammatory response linked to the stimulation of the secretion of cytokines and subsequent inflammatory mediators.71,72
4. Pharmacokinetics and biodistribution of nanoparticles
4.1. Opsonization and nanoparticles clearance
Whatever the route of exposure and absorption, once the nanoparticles reach the blood compartment, the nanoparticles are recognized by the immune system and thus are opsonized. This phenomenon is true for almost all types of nanoparticles, whether surface-modified or not, although the kinetics of the process may vary. Opsonization is a process of adsorption of plasma proteins onto the nanoparticles surface, making the particles prone to the processing by the immune system and/or modifies their biodistribution. Two types of opsonins, i.e., immune and non-immune opsonins interact with the nanoparticles in the blood. The immune opsonins are those that recognize nanoparticles as foreign objects and direct those to the immune system (i.e., to macrophages for phagocytosis), and comprise of complement proteins (different sub-classes of immunoglobulins, e.g. IgG, IgM) and complement-related proteins (e.g. C-reactive protein, serum amyloid protein, mannose-binding protein). The non-immune opsonins comprising of albumin, fibronectin, apolipoproteins, etc. are those that act as ligands and thus modify the distribution of nanoparticles by interacting with specific receptors on the cells.73Binding of opsonins onto the nanoparticles surface may be facilitated by one or various types of forces, such as, van der waals, electrostatic, hydrophilic/hydrophobic interactions.57 Opsonin binding often determines the biodistribution and fate of the nanoparticles. The opsonized nanoparticles are recognized and captured by the resident macrophages of the organs of reticuloendothelial system (RES) (e.g. liver and spleen), and thereby the nanoparticles are cleared from the blood compartment and are accumulated in the RES organs. The nanoparticles capture by the macrophages occurs either by the recognition of the nanoparticle-bound opsonins by the specific phagocytic receptors expressed on macrophages or by the non-specific adsorption process.57 Such accumulation of the nanoparticles in the RES organs may be undesired or desired depending on the type of the intended application of nanoparticles. For instance, the passive accumulation of nanoparticles in liver tissue may be beneficial for delivering therapeutic substances or imaging agents to the liver parenchyma and hepatocytes, while the accumulation in liver macrophages may be beneficial for delivering drugs to the macrophages for the treatment of macrophage-hosted diseases.74
Nanoparticle clearance from the systemic circulation and from the tissues may also be impacted by the balance in the T-helper type 1 and type 2 (Th1 and Th2) cell responses exhibited by the T-lymphocytes. These responses lead to the secretion of different sets of cytokines and chemokines which possess the ability to polarize the macrophages to M1 or M2 phenotypes. Th1 responses induce polarization of macrophages to M1 phenotype possessing slower nanoparticle clearance property, whereas, the Th2 responses induce M2 phenotypic macrophages possessing rapid nanoparticle clearance property,75,76 because the M2 macrophages express higher levels of different scavenger and lectin receptors compared to M1 phenotypic macrophages. Thus, the immune status of the subject and its impact on the nanoparticle clearance should be taken into account for assessing the nanoparticles pharmacokinetics and distribution and for interpretation of the in vivo data.
Opsonization-mediated systemic clearance of the nanoparticles has been shown to be minimized considerably by surface coverage of the nanoparticles with hydrophilic polymers. Coating of the nanoparticles surface with hydrophilic non-ionic polymer poly(ethylene glycol) (PEG), has been widely demonstrated to minimize the opsonization of the nanoparticles and thereby enhance their systemic longevity.77,78 The length and density of PEG chains on the nanoparticles surface was shown to regulate the opsonin binding by steric repulsion of the proteins approaching the nanoparticles surface.79,80 PEG molecular weights starting from 2 kDa are considered to be suitable to achieve the nanoparticles with prolonged systemic circulation time (also termed as ‘long circulating nanoparticles’).81 The concept of PEGylated long circulating nanoobjects was successfully developed which led to the commercialization of PEGylated liposomal doxorubicin (Doxil®/Caelyx®), while the docetaxel-loaded polymeric nanoparticle formulation BIND-014 is currently in phase II clinical testing for oncology application.
PEG has been widely considered as an inert polymer with non-immunogenic nature. However, the immunogenic property of PEG and its ability to stimulate the induction of anti-PEG antibodies, and the influence of these antibodies on the clearance of subsequent doses (doses following first injection) of PEGylated liposomes,82 polymeric nanoparticles,83 conjugates84 (generally termed as ‘accelerated blood clearance’ (ABC) phenomenon) compromizing the product efficacy has been the topic of frequent debate and controversy since several years. While the immunogenicity of PEG has been frequently demonstrated in various publications, both in animals and in humans,85,86 some recent studies have argued that a majority of the assays reported for anti-PEG antibodies may be flawed and lack specificity which urged the need of designing standard assays.87
The hypothesized mechanism of ABC phenomenon of PEGylated particles involved the production of anti-PEG IgM in the spleen, in a T-cell independent manner by directly activating the marginal zone B-cells, and selective binding of anti-PEG IgM on to the PEG of subsequent doses of the PEGylated particles administered into the body.88 In case of PEGylated liposomes, it was hypothesized that anti-PEG antibodies lead to complement activation and induce opsonization of the subsequent doses of the PEG liposomes administered into the body and/or inducing leakage of the liposome leading to the release of the encapsulated payload.89 Interestingly, however, it has been found that the physicochemical characteristics of PEGylated systems, and the time spacing between the two administered doses, and the diameter of nanoparticles, influenced the ABC phenomenon. For instance, the methoxy-PEG in the formulation induced high levels of anti-PEG antibodies compared to the hydroxy-PEG,90 and the nanoparticles of smaller diameter induced lower levels of antibodies as compared to the bigger particles (70 nm particles vs. 120 nm particles), and maintaining a spacing of at least 2 weeks between two injections resulted in overcoming the ABC phenomenon issue associated with the PEGylated nanoparticle products.91 Thus, in light of the previous reports on PEG's potential immunogenicity, the impact of physicochemical characteristics of PEG and PEGylated particles, and the time spacing between injections, the PEGylated nanoparticles should be appropriately characterized, and detailed in vitro and in vivo assessment of the safety aspects of PEGylated nanoparticles should be considered.
4.2. Nanoparticles biodistribution driven majorly by their physicochemical characteristics
The nanoparticles biodistribution in the body is dependent upon their physicochemical characteristics, such as, particle size and dispersity, surface charge, shape, rigidity, and composition.The nanoparticles, if aggregate into bigger sized particles during the course of their circulation in blood, are then filtered mechanically by the pulmonary capillaries and thus are retained in the lung tissue. Thus, the size stability of nanoparticles is essential to minimize such accumulation, if at all it is undesired. The nanoparticles of sizes smaller than 10 nm diameter are filtered by the kidneys and are subsequently excreted from the body, while the particles bigger than 10 nm diameter distribute to the kidney tissue.97,98
Furthermore, using nanodiamond particles, it has been shown in vitro that the nanoparticles possessing sharp shapes such as those having sharp corners and edges, irrespective of the size, surface properties and composition, pierced through the endosomal membranes in the hepatic carcinoma cells and trafficked into the cytoplasm. These nanoparticles in turn showed prolonged cytoplasmic residence and thus a reduced elimination from the cells.108 On the other hand, the cellular dynamics of spherical particles were different since they pierced less efficiently through the endosomal membrane and as a result persisted inside the endosomes. These particles then evolved with the endosomal maturation and subsequently eliminated rapidly from the cells via exocytosis.109
Shape and morphology of nanoparticles has been also shown to dictate the nanoparticles biodistribution to tissues in vivo and their penetration within the tissue, following intravenous injection in tumor bearing mice. In fact, the spherical-shaped and disc-shaped nanoparticles exhibited significantly higher distribution to the tumor tissue as compared to that of the nanoparticles possessing rod-shape and cage-shape, whereas, the rod-shaped and cage-shaped nanoparticles penetrated efficiently within the tumor tissue unlike that of the spherical-shaped and disc-shaped particles which remained mainly at the tumor periphery.110 Thus, the physicochemical characteristics of the nanoparticles responsible for their biophysical behaviour are crucial to understand and control, from the pharmacokinetic and biodistribution standpoint.
The length of acyl chains of PEG-lipids used in lipid nanoparticles composition influenced the kinetic of PEG desorption from the nanoparticle surface, and consequently, a different in vivo systemic clearance rates were found with nanoparticles containing PEG-lipids having different acyl chain lengths, when injected intravenously into mice.117 The nanoparticles containing PEG-lipids with short C14 acyl chains showed a systemic half-life of 5–6 h owing to a rapid desorption of PEG chains from the particle surface, whereas, the nanoparticles with long C20 acyl chains showed systemic half-life up to 10–12 h due to a slow desorption of PEG chains from the nanoparticle surface, indicating the impact of the nature of PEG-lipids on the pharmacokinetics of nanoparticles.
In case of polyethylene glycol–polylactic acid (mPEG–PLA) polymeric nanoparticles, alteration of the ratio of hydrophilic component PEG to the total mass of the copolymer in nanoparticles composition was shown to result in nanoparticles of different structures such as micelles or solid nanoparticle aggregates or vesicles.118 Such differences in structural properties may influence the nanoparticles in vivo behavior. For instance, when injected intravenously in preclinical models, the mPEG–PLA nanoparticles made using polylactic acid of small molecular weight (2 kDa) were found rapidly eliminated from blood circulation after few minutes of administration due to poor stability and subsequent degradation to PEG and PLA chains. On the opposite, the nanoparticles made from large molecular weight polylactic acid (30 kDa) were more stable and showed prolonged systemic circulation.81,119 Additionally, it was shown that the polymeric nanoparticles compositions containing varying PEG content or PEG lengths showed different plasma protein adsorption profiles when incubated in vitro with plasma.80 Nanoparticles with 5% PEG content adsorbed ∼3-folds lower quantity of plasma proteins compared to that of nanoparticles with 2% PEG content, while variation in PEG lengths had resulted in differences in the types of proteins adsorbed onto the nanoparticles.
The nanoparticles compositions containing small quantities of residual stabilizers used for the manufacture were shown to impact the nanoparticles interaction with the cells. For instance, the presence of residual stabilizer polyvinyl alcohol in PLGA nanoparticles composition was shown to negatively impact the cellular uptake of these nanoparticles in vitro.120 Overall, the potential impact of the differences in the composition of nanoparticles on their interaction with biological milieu and consequently on their in vivo behavior should be taken into account during their biopharmaceutical assessment.
5. Nanoparticles toxicity assessment: in vitro
It is clear from that described in the previous section that the physicochemical characteristics of the nanoparticles, such as, influence their biophysical properties. The nanoparticles designed using different raw materials which exhibit different chemical compositions, and those designed using different manufacturing processes potentially exhibit different physicochemical characteristics. Such differences may result in altered nano–bio interactions which may have impact on performance and safety of the nanoparticles. Thus, for any new type of nanomaterial developed for biomedical applications, a detailed understanding of the nano–bio interactions, the toxicological properties, and the associated mechanisms, using appropriate tools and methodologies is crucial for anticipation of its performance and safety.Especially, the safety assessment of the nanoparticles should be performed using both in vitro and in vivo models. In vitro assessment has certain advantages over in vivo assessment, such as, low quantities of samples needed for testing, rapidity, lower cost, better control on variability, allows studying the mechanistic aspects, and minimizes the use of/sacrificing of laboratory animals. The in vitro studies including mechanistic understanding of the nano–bio interactions, both at molecular and genetic level, undoubtedly serve during the interpretation of in vivo preclinical data and subsequently for the interpretation of the clinical data. Despite of the above advantages, the in vitro assays cannot be considered as a standalone or substitute to the in vivo assays which allow the real determination of the toxicity in the complex biological environment, because of the challenges associated with simulating in vivo conditions in in vitro models. Also, in in vitro conditions, the nanoparticles are in direct contact with the cells and thus remain as a reservoir at higher concentrations close to the cells, whereas, in vivo the nanoparticles distribute throughout the body and the fraction of nanoparticles reaching the cells may not be as dramatic as that happens in vitro. Thus, appropriate attention is needed during the interpretation of the in vitro toxicity results. In this section, the in vitro assays potentially useful for the assessment of nanoparticles toxicity, their principles, methodologies, benefits and challenges wherever relevant, are described (see Fig. 2).
 | ||
| Fig. 2 Schematic representation of the toxicity assessment of nanoparticles, in vitro and in vivo. TB: trypan blue; PI: propidium iodide; NR: neutral red; CAE-EHD-1: calcein acetoxymethyl ester/ethidium homodimer-1; LDH-lactate dehydrogenase; Annexin V-FITC/PI: Annexin V–fluorescein isothiocyanate/propidium iodide; TUNEL-terminal deoxynucleotidyl transferase mediated dUTP-biotin nick end labeling; ROS: reactive oxygen species; DCFH: 2,7-dichlorodihydrofluorescein; EPR: electroparamagnetic resonance; SOD: superoxide dismutase; CAT: catalase; GSH-glutathione; GPx: glutathione peroxidase; GR: glutathione reductase; GST: glutathione-S-transferase; LPO: lipid peroxidation; 8-OHdG: 8-hydroxyl-2′-deoxyguanosine; NPs: nanoparticles; NBA: Northern blot analysis; RPA: ribonuclease protection assay; qRT-PCR: quantitative real-time polymerase chain reaction; HES: hematoxylin eosin and saffran. | ||
The main components of the body that are often exposed to nanoparticles, after any route of administration or exposure, are the blood components and the cells/tissues. Thus, the in vitro toxicity assessment is generally performed to understand the nanoparticle–biological interactions and safety at the cellular/sub-cellular level and/or at the level of blood components.
5.1. Interaction with cells
This section provides details of the principles and methodologies of various in vitro assays used for the toxicity assessment of nanoparticle systems.5.1.2.1. Tetrazolium salts assay. This assay involving the use of the tetrazolium salts, such as, MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) or MTT (3-(4, 5- dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) which are reduced intracellularly by the living cells to produce formazan dyes (Fig. 3) whose absorbance can be quantified using spectroscopy, is the most widely used assay for in vitro cell toxicity assessment. Mitochondrial succinate dehydrogenases of the living cells bioreduce the incubated soluble tetrazolium salts to the insoluble purple coloured formazan crystals which are impermeable through the cell membrane. These insoluble crystals accumulate within the healthy cells,121 are then solubilized by the addition of solvents such as DMSO (Dimethyl sulfoxide) or detergents (e.g. sodium lauryl sulfate), and the absorbance of the resultant colour is measured using UV-visible spectrophotometer.122,123 The percentage of surviving cells can be calculated as the absorbance ratio of the treated cells to that of the untreated cells.
 | ||
| Fig. 3 Intracellular reduction of MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) to purple coloured formazan dye. | ||
The benefits of this assay compared to other toxicity assays include simplicity, rapidity, and the need of simple optical density acquisition,124 while the drawbacks include the inefficiency of some human cell lines at processing tetrazolium salts. Additionally, the changes in the pH of the culture medium, culture media supplements such as serum, cholesterol, ascorbate125 etc. may alter the measurements and thus needs particular attention during interpretation of the assay results.
5.1.2.2. Alamar blue assay. This assay is a cell viability indicator measuring the reductive environment in the cell cytoplasm during cell metabolism, which is measured spectrophotometrically through the conversion of fluorimetric/colorimetric redox indicators. Upon incubation of the cells with alamar blue, the metabolic activity of cells reduces alamar blue (resazurin, the oxidized form), a non-toxic and non-fluorescent cell permeable product, to resorufin (the reduced form), a bright red fluorescence product. The resultant fluorescence can be measured at 590 nm126 (Fig. 4), which reflects the viable cell number and changes in the cellular redox activity.127 This assay was used to determine the cell viability of cobalt–ferrite nanoparticles using normal mouse dendritic cells and also a variety of cancer cell lines.128
 | ||
| Fig. 4 Reduction in viable cells, of non-fluorescent alamar blue (resazurin) to a bright red fluorescent resorufin. | ||
The disadvantage of this assay is that it is not a direct cell counting technique, and sometimes a false change in fluorescence may result due to auto-reduction of resazurin. The auto-reduction can be inhibited by incorporating suitable redox stabilizing agents (e.g. potassium ferrocyanide, ferric salt, ferricinium) in the control and the test samples. An unintended reduction of resorufin to dihydroresorufin which is a non-colored and non-fluorescent product may also occur resulting in the loss of the desired end product. The formation of dihydroresorufin can be inhibited by addition of poising agents such as methylene blue, toluidine blue, azure I and gallocyanide in the concentrations sufficient to maintain the potential of the growth medium above −0.1 volts.129
5.1.2.3. [3H]-Thymidine incorporation into the newly synthesized cellular DNA. Uptake of [3H]-thymidine into newly synthesized DNA during S-phase (synthesis phase) of the cell cycle is a sensitive measurement of the cell proliferation. Following treatment of cells with the nanoparticles whose toxicity is to be assessed, the cells are isolated and incubated with [3H]-TdR. After pre-determined incubation time, the cells are washed to remove the un-incorporated label, while the label incorporated into cellular DNA is measured using scintillation counter. The drawbacks associated with this method include the cost of the radioactive material, and also the need of special training and the approved facility to handle the radioactive materials.130 Moreover, the radioactive isotope 3H in [3H]-TdR was shown to inhibit to a certain extent the rate of DNA synthesis thereby potentially interfering with the assessment, and thus the use of non-radioactive stable isotopes instead of radioactive isotope for such purpose has been suggested.131
5.1.2.4. Clonogenic assay. Clonogenic assay, also termed as colony forming efficiency (CFE) assay, can be used to study the impact of the nanoparticle samples on the cell survival and proliferation over extended periods of time (up to several weeks). The methodology involves incubation of cells with nanoparticles sample whose impact on the cells is to be assessed, followed by staining of the cells with crystal violet or nuclear stains, and counting the colonies of the proliferating cells by visual observation.132 This method has been successfully employed for the assessment of the effects of carbon nanotubes on human bronchial epithelial and on human keratinocyte cell lines.133 Interestingly, it was observed that increasing doses of the nanotubes resulted in decreased number of colonies, suggesting a dose-dependent toxicity of the nanotubes in vitro.
5.1.3.1. Trypan blue assay. Trypan blue assay is based on the principle that the live cells possessing intact cell membrane excludes trypan blue, a negatively charged dye, whereas, the dead cells are permeable to the dye and thus take up the dye resulting in a strong absorbance at 605 nm. In this assay, the cell suspension pre-treated with nanoparticle sample whose impact is to be assessed is incubated with trypan blue, and is then visually examined using microscopy to determine whether the cells take up or exclude the dye. Viable cells appear not colored as these cells do not take up the dye, whereas, dead cells take up the dye into the cytoplasm and thus appear blue in colour.138 The number of viable cells, an increase or decrease, in comparison to the untreated cells, is counted and the ratio is determined. The advantages of this assay include simplicity and low cost, while the drawbacks include potential variability associated with the cell counting apparatus such as hemocytometer,139 and also that even the living cells take up the dye if incubated with the dye for longer durations. For instance, trypan blue assay has been reported for the assessment of cytotoxicity of the nanocrystalline magnesium ferrites (MgFe2O4) of about 20 nm diameter on the MCF-7 breast cancer cell line.140
5.1.3.2. Propidium iodide assay. Propidium iodide is a negatively charged DNA intercalating fluorescent agent employed to analyze the cell cycle events using flow cytometric measurements of cellular DNA. Propidium iodide does not permeate through the viable cell membranes and thus is excluded by the viable cells. In the permeable cells, propidium iodide binds to and intercalates with DNA and RNA thereby staining these nucleic acids whose content could be measured using flow cytometry. Thus, for specific DNA analysis, the samples are treated with ribonuclease to eliminate RNA. The methodology involves incubation of the cells with the nanoparticles samples whose toxicity assessment is intended. After the predetermined interval, the cells are washed with pH 7.4 phosphate buffered saline (PBS). About 1 × 106 cells are incubated overnight at 4 °C in 70% ethanol to fix and permeabilize the cells, and subsequently centrifuged to remove ethanol, washed with PBS, and treated with extraction buffer (a mixture of 192 parts of 0.2 M disodium hydrogen phosphate and 8 parts of 0.1 M citric acid). Then the cells are washed with PBS and treated with deoxyribonuclease-free ribonuclease-A (200 μg ml−1) for 30 min at 37 °C to inactivate RNA to be able to measure DNA. Then the cells are stained with propidium iodide (50 μg ml−1) in PBS for about 10 min followed by the measurement using a flow cytometer.141 Quantification of the cellular DNA content using this assay provides information such as the identification of the cells in various phases of cell cycle, the DNA damage, and the measurement of the apoptotic cells.
5.1.3.3. Neutral red assay. The principle of neutral red assay is that the viable cells take up the neutral red dye (3-amino-7-dimethylamino-2-methylphenazine hydrochloride), which is unionized at physiological pH, by active transport and incorporate into the intracellular lysosomes where the dye gets protonated (Fig. 5) and thus accumulate, while the dead cells do not take up the dye. The methodology involves the incubation of cell suspension post-treatment with the test sample whose toxicity testing is intended, with the neutral red dye for 2 to 4 h, and subsequently washing the cells in PBS followed by extraction and spectrophotometric quantification of the incorporated dye. This assay is cost-effective and more sensitive than other cytotoxicity assays such as tetrazolium salts assay,142 while the potential drawback is the impact on the dye quantification results by the agents that affect the lysosomes within the cells where the dye is retained.
 | ||
| Fig. 5 Chemical structures of neutral red as neutral form and acidic form. | ||
5.1.3.4. Calcein acetoxymethyl ester/ethidium homodimer assay. This assay involving the labeling of living cells is based on the principle of the enzymatic conversion of virtually non-fluorescent cell-permeable calcein violet acetoxymethylester (obtained by the modification of anionic carboxylic acid functions of calcein violet with the acetoxymethylester groups) to the intensely fluorescent anionic calcein violet (λex at 400 nm and λem at 452 nm) by the intracellular esterases by hydrolysis in the living cells143,144 (Fig. 6). Intracellular esterases cleave the parent compound to result in an anionic fluorescent dye calcein violet which is retained in the cells to a much greater extent than its uncharged parent compound. The methodology involves the incubation of cell suspension treated with the test sample whose toxicity assessment is intended, with the calcein violet acetoxymethylester solution (whose stock solution is prepared in anhydrous dimethylsulfoxide to avoid hydrolysis of the compound) for about 30 minutes. The cells are centrifuged, washed once with PBS, and are observed either using fluorescence microscope or are analyzed using flow cytometer.
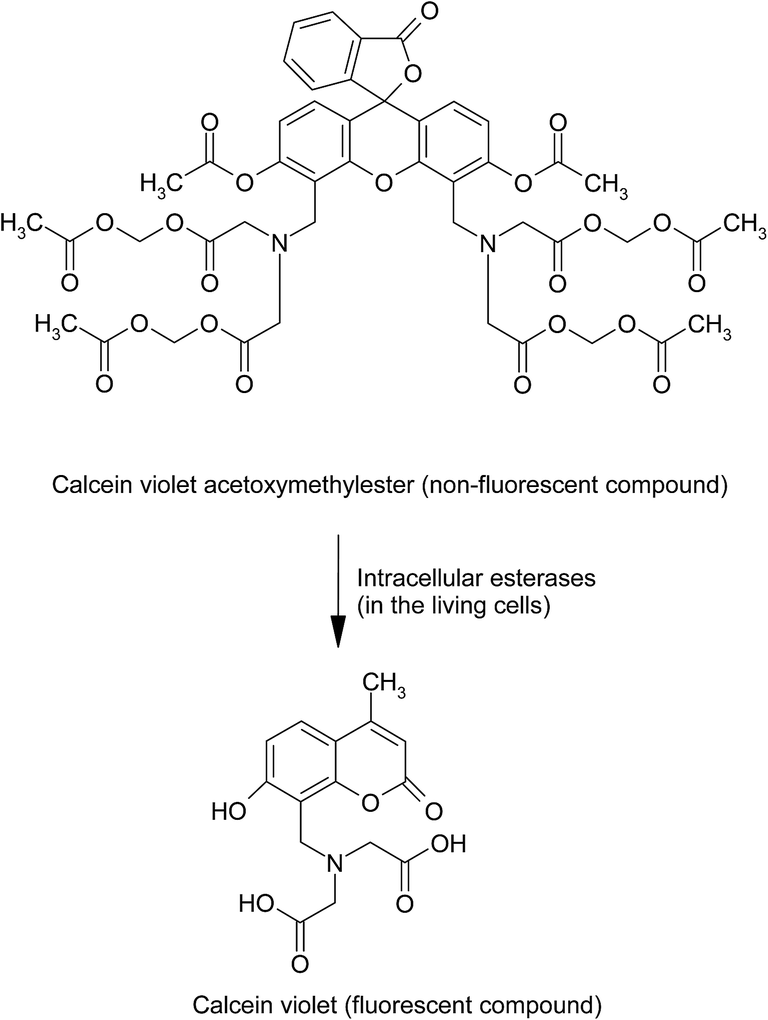 | ||
| Fig. 6 Intracellular conversion of non-fluorescent calcein violet acetoxymethylester to the fluorescent anionic calcein violet by esterases in the living cells. | ||
Ethidium homodimer (EthD-1) (5,5′-[1,2-ethanediylbis(imino-3,1-propanediyl)]bis(3,8-diamino-6-phenyl) dichloride dihydrochloride) is a high-affinity nucleic acid stain which exhibits 40-folds enhancement in the fluorescence after binding to the cellular DNA (λex at 528 nm/λem at 617 nm)145 (Fig. 7). When incubated with the cell suspension, EthD-1 enters the cells possessing damaged membranes and exhibit enhanced bright red fluorescence that could be visualized using fluorescent microscope or by flow cytometer, but is excluded by the living cells possessing intact membrane.
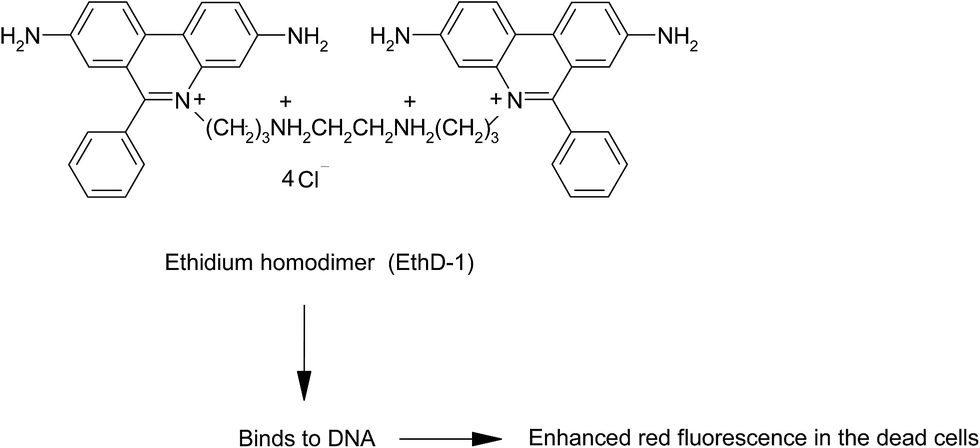 | ||
| Fig. 7 Ethidium homodimer (5,5′-[1,2-ethanediylbis(imino-3,1-propanediyl)]bis(3,8-diamino-6-phenyl) dichloride dihydrochloride) penetrate the dead cells and binds to DNA thereby exhibiting red fluorescence. | ||
A combined assay to determine both the living and the dead cell population in the cell suspension following treatment with the nanoparticles samples whose toxicity assessment is intended, could be carried out by incubating the cell suspension with both calcein violet acetoxymethylester which allows the measurement of living cell population and ethidium homodimer which allows the measurement of the dead cell population. This assay could be carried out using Live/Dead® viability/cytotoxicity kit (Molecular probes, Invitrogen technologies).146
5.1.3.5. Lactate dehydrogenase (LDH) assay. This assay could be used, to assess the cell membrane damage by determining the cytoplasmic LDH release into the medium following incubation with the nanoparticles sample or to assess the cell viability by lysing the membrane of the living cells using the detergent Triton X-100 and subsequently quantifying the total LDH levels. LDH is a stable cytoplasmic oxido-reductase enzyme that catalyzes the inter-conversion of pyruvate to lactate in the living cells (Fig. 8A). LDH catalyzes the reduction of nicotinamide adenine dinucleotide (NAD+) to NADH, which is subsequently used to stoichiometrically convert the INT ((2-(4-Iodophenyl)-3-(4-nitrophenyl)-5-phenyl-2H-tetrazolium chloride)) to a red colored soluble formazan product (Fig. 8B) whose absorbance can be measured spectrophotometrically at 490 nm. For example, the LDH assay was used to assess the toxicity of silver, molybdenum and aluminium nanoparticles possessing diameters ranging between 15 to 30 nm.147 The nanoparticles-treated cells showed considerable LDH leakage indicating plasma membrane damage and thus suggesting the nanoparticles-induced cell toxicity.
 | ||
| Fig. 8 Schematic representation of the formation of NADH in the living cells during lactate dehydrogenation (A), followed by the reaction between the so formed NADH and INT resulting in the formation of red colored INT formazan (B). | ||
 | ||
| Fig. 9 Illustration of apoptotic bodies in an apoptosis induced cell. | ||
5.1.4.1. Annexin V-FITC/propidium iodide assay. Phosphatidyl serine, a phospholipid, which is typically oriented toward the cytoplasmic side of the cell membrane of healthy cells, becomes exposed to the outer side of the cell membrane in the cells undergoing apoptosis.148 Thus, the expression of phosphatidyl serine on the cell membrane is considered as one of the hallmarks of the cellular apoptosis. Annexin V, a calcium dependent phospholipid-binding protein possessing a high affinity for phosphatidylserine, is employed in the form of conjugate with the fluorescent probe fluorescein isothiocyanate (FITC) for the determination of the presence of phosphatidylserine moieties on the cell membrane of apoptotic cells. Annexin V–FITC bound onto the apoptotic cells can be detected by fluorescence detection and can be quantitatively measured using flow cytometry.
A combination assay wherein the cells are treated with both Annexin V–FITC and propidium iodide allows the differentiation among the early apoptotic cells which are annexin V positive and propidium iodide negative, the late apoptotic cells which are annexin V positive and propidium iodide positive, and the viable cells which are negative to both annexin V and propidium iodide. For instance, the Annexin V-FITC/propidium iodide assay was employed to determine the in vitro toxicity and induction of apoptosis by silica nanoparticles possessing 20 nm diameter on human HepG2 hepatoma cells and normal human L-02 hepatic cell lines.149 The results revealed a dose-dependent induction of apoptosis by the nanoparticles, and the assay allowed distinguishing the early and the late phase apoptosis in the cells.
5.1.4.2. Microscopic assessment of apoptotic bodies. The morphological characteristics of the apoptotic cells, such as, condensation and marginalization of chromatin, fragmentation of nuclei, cell shrinkage can be identified by staining the cells with Hoechst stain (e.g. Hoechst 33258). The methodology involves treatment of the nanoparticles-treated cells fixed and placed on the glass slides with Hoechst stain dissolved in citric acid [0.01 M], disodium phosphate [0.45 M] buffer containing 0.05% Tween-20, followed by observation of the cells under fluorescent microscope150 and counting various apoptotic bodies for calculation of their percentage in comparison with the control cells (i.e. the cells not treated with nanoparticles sample).
5.1.4.3. DNA laddering/DNA fragmentation assay. DNA fragmentation, the cleavage of chromatin DNA into 180–200 bp oligonucleosomal units, is one of the hallmarks of apoptosis.151 When run on gel electrophoresis, the cleaved oligomers appear as a DNA ladder. In the cells undergoing apoptosis, caspase-3 (the member of cysteine–aspartic acid protease family) initiates the DNA fragmentation by proteolytic inactivation of the inhibitor of caspase activated deoxyribonuclease (ICAD), resulting in the release of the endonuclease, i.e., caspase activated deoxyribonuclease (CAD) that causes DNA fragmentation.152 The assay methodology involves subjecting the cells treated with nanoparticles whose toxicity assessment is to be done, to the DNA extraction and isolation, followed by resolving the isolated DNA on 1.5% agarose gel containing 3 μg ml−1 ethidium bromide and subsequently visualizing the bands using a UV transilluminator.153 Alternatively, the cells could be lysed using the DNA fragmentation lysis buffer (0.1% Triton X-100, 5 mM Tris–HCl, pH 8.0, 20 mM EDTA), followed by the selective precipitation of the unfragmented, high-molecular weight DNA using polyethylene glycol 8000, while the fragmented DNA remaining in the supernatant can be directly analysed using agarose gel electrophoresis or using the fluorescent dye Hoechst 33258.154
For instance, silver nanoparticles when incubated with HT-1080 Human fibrosarcoma and A431 Human skin carcinoma cells induced apoptosis as observed from the oligonucleosomal DNA fragments or DNA laddering at the nanoparticles concentration of 6.25 μg ml−1 indicating the nanoparticles toxicity.153
5.1.4.4. Comet assay or single cell gel electrophoresis. DNA damage in the cells, if any, resulting from the exposure to nanoparticles could be determined using comet assay. In this assay, the cells pre-incubated with test sample whose toxicity assessment is intended, are lysed to remove proteins, and the DNA is denatured under alkaline or neutral conditions, stained with ethidium bromide and subjected to electrophoresis to observe the broken DNA fragments or damaged DNA portions. The degree of DNA damage is detected by the extent of tailing (appear as a comet). Using this assay, the kinetics of the progression of DNA fragmentation could also be elaborated.155 The main drawback of comet assay is its inability to measure fixed mutations.
5.1.4.5. TUNEL (terminal deoxynucleotidyl transferase mediated dUTP-biotin nick end labeling) assay. During apoptosis, the chromatin structure of the cellular DNA is degraded into fragments of 50–300 kilobases and small oligomers of about 200 bp with a large number of 31-OH ends exposed. TUNEL assay is used to detect the DNA fragmentation by labeling the DNA strand breaks. The 3′-OH termini exposed due to the fragmentation of genomic DNA are labeled with the fluorochrome-labeled dUTP (e.g. Br-dUTP) with the aid of terminal deoxynucleotidyl transferase, an exogeneous DNA polymerase-I which repairs isolated DNA fragments, leading to the formation of the low molecular weight double-stranded DNA and the high molecular weight single stranded DNA.156 The DNA analysis could then be carried out either by using flow cytometry or by image analysis.
TUNEL assay has been employed for the evaluation of the toxicity of metal oxide nanoparticles, such as, copper oxide (CuO) and silica (SiO2) on human adenocarcinoma A549 cell line.157 When the cells were incubated with these nanoparticles at the concentrations of 30 μg ml−1 for 8 h, no significant differences between the control and the nanoparticles-treated cells were observed suggesting the lack of toxicity by these nanoparticles at the concentration tested.
5.1.4.6. Assay of mitochondria-dependent apoptosis. Involvement of mitochondria in apoptosis could be assessed by examining various aspects, importantly the measurement of the overexpression of Bax and cytochrome-C, and the caspase-3 cleavage analysis.
Bax, an important pro-apoptotic protein of the Bcl-2 family, can translocate to the outer mitochondrial membrane and insert into the mitochondria and form oligomers contributing to the formation of mitochondrial permeability transition pore (PTP). Opening of the mitochondrial PTP can lead to the release of cytochrome C (cyt-C) into the cytoplasm which is a key indicator of the mitochondrial dependent apoptosis pathway. Methodologically, the cells are incubated with nanoparticles for pre-determined time interval, followed by harvesting about 1 × 107 cells. The proteins Bax and Cyt-C are then extracted from the cells using protein extraction kit and are subsequently quantified using BCA protein assay kit. Protein samples are then resolved using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and are subsequently transferred onto the nitrocellulose membranes. The membranes are washed, incubated with anti-Bax antibody and anti-cytochrome-C antibody at 4 °C overnight. Immunodetection is then performed with secondary horse radish peroxidase (HRP)-conjugated antibody.158
5.1.4.7. Caspase-3 assay. Caspases (cysteine-requiring aspartate proteases) are a family of proteases which are important entities in the process of apoptosis. Caspase-3, a member of CED-3 (Caenorhabditis elegans gene ced-3) subfamily of caspases, is one of the critical enzymes of apoptosis. It can process procaspases and specifically cleave most of the caspase-related substrates including many key proteins involved in apoptosis regulation159 leading to cell death. In addition, caspase-3 plays an important role in mediating nuclear apoptosis including chromatin condensation, DNA fragmentation and cell blebbing.160 The activity of caspase-3, if elevated in the cells as a result of nanoparticles treatment, can be determined using caspase-3/CPP32 colorimetric assay kit. This assay is based on the hydrolysis of the peptide substrate Asp-Glu-Val-Asp p-nitroanilide (DEVD-pNA) by casapse-3, leading to the release of p-nitroaniline which is assessed spectrophotometrically at 405 nm.
5.1.5.1. Measurement of ROS. Nanoparticles toxicity may disturb the oxidative balance of the cells resulting in the production of abnormally elevated concentrations of ROS161 (e.g. superoxide anion (O2−), free radicals (e.g. hydroxyl radical (OH˙), peroxy radical (ROO˙) and hydrogen peroxide (H2O2)) or reactive nitrogen species (RNS) (e.g. nitric oxide (NO˙), peroxynitrite ion (ONOO−), peroxy nitrous acid (ONOOH)). ROS and RNS exhibit toxic effects by damaging the DNA, proteins and lipids in the cells resulting in the abnormal cellular function. The cellular ROS could be measured using various methods differing in specificity, sensitivity, and the ability to measure intracellular and/or extracellular ROS.
5.1.5.1.1. DCFH (2,7-dichlorodihydrofluorescein) assay. In this assay, 2,7-dichlorofluorescein diacetate (DCFH-DA), a non fluorescent lipophilic probe is used. The diacetate moiety provides lipophilicity to the molecule. Intracellularly, DCFH-DA is hydrolyzed by esterases to the impermeable non-fluorescent reduced DCFH (2′,7′-dichlorofluorescein) which is rapidly oxidized by the intracellular ROS to the highly fluorescent 2′,7′-dichlorofluorescein (DCF) (Fig. 10). The fluorescence of the resultant DCF could be measured at λex of 485 nm and λem 520 nm,162 wherein the measured fluorescence intensity is proportional to the ROS levels in the cytoplasm.
 | ||
| Fig. 10 Structural representation of the conversion of the reagent DCFH-DA (2′,7′-dichlorofluorescein diacetate) to DCFH in the cells by esterases, and subsequent reaction of DCFH with intracellular reactive oxygen species resulting in the formation of fluorescent DCF (2′,7′-dichlorofluorescein). | ||
Briefly, the methodology involves the incubation of test sample-treated cells with DCFH-DA solution (methanolic solution of DCFH-DA diluted in serum- and additive-free culture medium) at 37 °C for 30 min. The cells are then washed with PBS to remove excess DCFH-DA, lysed in alkaline solution, and then centrifuged for 10 min. The fluorescence intensity of DCF formed in the supernatant is measured at λex of 488 nm and λem of 525 nm.163,164
5.1.5.1.2. EPR (electroparamagnetic resonance) technique. EPR spectroscopy has been widely used for the assessment of nanoparticle induced ROS generation. It allows the identification and quantification of specific free radical generated by using specific spin traps or probes. For instance, the probe 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) which is specific for the formation of hydroxyl radical (DMPO reacts with the hydroxyl radicals and forms DMPO-OH)165 or 1-hydroxy-4-phosphonooxy-2,2,6,6-tetramethylpiperidine (PP-H) which is specific for the formation of superoxide anion166 are incubated with the cells treated with the test sample, and the supernatant is isolated and analyzed using EPR spectrophotometer.165,167 This method has been successfully employed for the cytotoxicity assessment of titanium dioxide nanoparticles of 100 nm size on human bronchial epithelial cells.168
5.1.5.1.3. Plasmid DNA scission assay. This assay has been used to assess ROS production in some studies169,170 by using circular bacterial plasmid DNA that is wound into supercoiled structure. Presence of ROS, particularly hydroxyl radicals, cleaves the bonds holding the supercoiled structure, thereby the circular structure becomes unwind and may become linear in the presence of high ROS content. These various forms of DNA could be distinguished by their electrophoretic mobility on agarose gel, wherein, the mobility of supercoiled structure is higher that that for the circular form. Ethidium bromide staining of the gel allows the quantification of the supercoiled band intensity whose depletion is a measure of plasmid damage by the ROS.171 This assay may not be considered as sensitive for nanoparticles testing because some fractions of DNA may potentially bind to the nanoparticles surface.
5.1.5.1.4. Oxidative stress assays. Oxidative stress is defined as the excess formation or inefficient removal of highly reactive species due to the imbalance created between pro-oxidants (e.g. ROS) and anti-oxidant defense mechanisms of the body.172 The oxidative stress markers include superoxide dismutase (SOD), catalase, glutathione, glutathione reductase, glutathione transferase, and lipid peroxidation. Of these, superoxide dismutase is an importance antioxidant enzyme which catalyses the dismutation of superoxide anion (O2˙−) into H2O2 and O2 providing an important defense against the oxidative damage. The levels of oxidative stress markers, in case of toxicity, may be either elevated or depleted.163,173–175
5.1.5.1.4.1. Measurement of superoxide dismutase activity. Various assays, such as, xanthine–xanthine oxidase assay (XOD) and nitroblue tetrazolium (NBT) assay have been employed for the indirect measurement of SOD activity.176 SOD activity in the experimental samples is measured as the percentage inhibition of the rate of formation of formazan dye. In xanthine–xanthine oxidase assay, the superoxide anions generated as a result of the conversion of xanthine and O2 into uric acid and H2O2 reduce the tetrazolium salt WST-1 into WST-1 formazan dye whose absorbance is measured at 450 nm. Addition of the test sample containing SOD to this reaction reduces superoxide anion levels, thereby lowering the rate of formation of formazan dye indicated by a decreased absorbance at 450 nm, which is a measure of the activity of SOD in the test sample. This method possesses drawbacks, such as, the poor water solubility of formazan dye and its reaction with the reduced form of xanthine oxidase.
In Nitroblue tetrazolium assay, the superoxide anions generated during autooxidation of the added hydroxylamine reagent, convert the water-soluble yellow coloured NBT to the blue coloured NBT-diformazan which can be measured spectrophotometrically at 560 nm. Presence of SOD in the test sample reduces the superoxide anion levels by converting it to H2O2 and molecular oxygen thereby lowering the rate of formation of diformazan dye.
5.1.5.1.4.2. Measurement of catalase activity. Catalase is another important antioxidant enzyme that catalyzes the decomposition of hydrogen peroxide into water and oxygen molecules.177 The presence of catalase (increased or decreased levels) in the test sample can be assessed by the addition of hydrogen peroxide. The methodology involves mixing of the cell lysate obtained from the cells treated with test sample, containing known amount of protein, with potassium phosphate buffer (pH 7.0) containing H2O2. The decrease in the absorbance of H2O2 is measured spectrophotometrically at 240 nm. Catalase activity is calculated from the slope of the H2O2 absorbance curve and normalized to the protein concentration.178
Catalase also exhibits peroxidase activity, in which low molecular weight alcohols such as methanol serve as electron donors. This assay is based on the principle that the enzyme catalyzes the conversion of methanol, in the presence of H2O2, to formaldehyde which then reacts with chromogenic substrate, 4-amino-3-hydrazino-5-mercapto-1,2,4-triazole (called as purpald) resulting in the formation of a colourless compound (Fig. 11). This compound upon oxidation forms a purple coloured product which can be spectrophotometrically measured.
 | ||
| Fig. 11 Schematic representation of the formation of formaldehyde from methanol by the action of catalase, and the interaction of so formed formaldehyde with the colorless 4-amino-3-hydrazino-5-mercapto-1,2,4-triazole (purpald) resulting in the formation of a colored oxidation end product. | ||
5.1.5.1.4.3. Measurement of glutathione (GSH) activity. GSH is an antioxidant present in the cells, whose functional role is to detoxify the ROS and hence essential in maintaining the reduced environment in the cells.
| R˙ + GSH → RH + GS˙ |
| GS˙ + GS− → GSSG˙− |
| GSSG˙− + O2 → GSSG + O2˙− |
At high concentrations of ROS exposure, an imbalance between the levels of oxidized glutathione (GSSG) and reduced glutathione (GSH) is observed. Such changes in GSH:GSSG ratio is an indicator of oxidative stress which can be assessed using liquid chromatographic methods.179 Appropriate care should be exercised when using this method because during chromatographic estimation process, autooxidation may potentially occur leading to the overestimation of GSSG.
One of the frequently used methods to measure GSH is the O-phthaldialdehyde (OPA) method. OPA is non-fluorescent and reacts with sulfahydril and primary amino groups of GSH to result in the formation of highly fluorescent iso-indole adducts (OPA-GSH adducts) (Fig. 12A).
 | ||
| Fig. 12 Structural representation of (A) O-phthaldialdehyde method and (B) DTNB (5,5-dithio-bis(2-nitro benzoic acid) method for the measurement of glutathione activity. | ||
GSH can also be measured by using DTNB (5,5-dithio-bis(2-nitro benzoic acid), Ellman's reagent)180 in presence of NADH which facilities the reduction of GSSG to GSH by glutathione reductase. The sulfahydril group of GSH subsequently reacts with DTNB resulting in the formation of yellow colored 5-thio-2-nitro benzoic acid (TNB) (Fig. 12B) which can be measured colorimetrically at 412 nm to obtain the concentration of GSH.
5.1.5.1.4.4. Measurement of glutathione peroxidase (GPx) activity. Glutathione peroxidase is an antioxidant enzyme which catalyses the detoxification of H2O2 and lipid hydroperoxides by GSH181 thereby protecting the cells against oxidative damage.


Glutathione peroxidase activity can be assessed using lipid hydroperoxide substrate tertiary-butyl hydroperoxide.182 In this method, the presence of GPx in the cell lysate catalyses the detoxification of t-butyl hydroperoxide by GSH (reduced form) resulting in the formation of GSSG (the oxidized glutathione) which is subsequently recycled to GSH by glutathione reductase in the presence of NADPH present in the reaction mixture.

The methodology involves the addition of reaction mixture containing t-butyl hydroperoxide (30 mM), reduced GSH (2 mM), GPx (0.5 unit ml−1) and NADPH (0.25 mM) to the cell lysate, followed by measuring the decrease in NADPH absorbance for 3 min spectrophotometrically at 340 nm. GPx activity is calculated from the NADPH absorbance standard curve.
5.1.5.1.4.5. Measurement of glutathione reductase (GR) activity. Glutathione reductase catalyses the recycling of GSH from GSSG in presence of NADPH, thus offering protection against the oxidative stress. GR activity is measured by the addition of the reaction mixture containing 0.1 M phosphate buffer (pH 7.4), 0.66 mM GSSG and 0.1 mM NADPH to the cell lysate, and any decrease in NADPH absorbance can be measured spectrophotometrically at 340 nm. GR activity is calculated from the NADPH absorbance standard curve.183
5.1.5.1.4.6. Measurement of glutathione-S-transferases (GSTs) activity. Glutathione transferases are antioxidant enzymes which catalyze the conjugation of electrophilic substrates to GSH, and thus are involved in the detoxification process.184 GST activity is assessed from its ability to mediate the conjugation of GSH with CDNB (1-chloro-2,4-dinitrobenzene), wherein the extent of conjugation leads to proportional change in the absorbance measured at 340 nm. The methodology involves the addition of the reaction mixture containing 1 mM GSH, 1 mM CDNB and 0.1 M potassium phosphate (pH 6.5) to the cell lysate, followed by the measurement of absorbance spectrophotometrically at 340 nm.185
5.1.5.1.4.7. Measurement of lipid peroxidation (LPO). LPO is defined as the oxidative deterioration of lipids containing C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. LPO causes modification in the permeability and fluidity of membranes of mitochondria and lysosomes resulting in damage to these membranes. In case nanoparticles induce the oxidative stress in cells, the polyunsaturated fatty acids in the cellular lipid membrane may undergo peroxidation resulting in the formation of unstable lipid hydroperoxides which subsequently decompose into lipid peroxidation products, such as, malondialdehyde (MDA), 4-hydroxynoneal (4-HNE). In the presence of 2-thiobarbutyric acid (TBA), MDA reacts and leads to the formation of a red colored 1:2 MDA:TBA adduct which can be quantified colorimetrically to assess the extent of lipid peroxidation (Fig. 13).
C bonds. LPO causes modification in the permeability and fluidity of membranes of mitochondria and lysosomes resulting in damage to these membranes. In case nanoparticles induce the oxidative stress in cells, the polyunsaturated fatty acids in the cellular lipid membrane may undergo peroxidation resulting in the formation of unstable lipid hydroperoxides which subsequently decompose into lipid peroxidation products, such as, malondialdehyde (MDA), 4-hydroxynoneal (4-HNE). In the presence of 2-thiobarbutyric acid (TBA), MDA reacts and leads to the formation of a red colored 1:2 MDA:TBA adduct which can be quantified colorimetrically to assess the extent of lipid peroxidation (Fig. 13).
 | ||
| Fig. 13 Structural representation of the assessment of lipid peroxidation by reacting the lipid peroxidation product malondialdehyde with 2-thiobarbutyric acid to result in the formation of red coloured adduct which can be quantified using colorimetry. | ||
The methodology involves the incubation of 200 μl of cell suspension with 800 μl of reaction mixture containing TBA (0.4% w/v), SDS (0.5% w/v) and acetic acid (5% v/v, pH 3.5) for 1 h at 95 °C. The sample is cooled and centrifuged at 5000 rpm for 5 min, and the absorbance of the supernatant is measured at 532 nm. The result is expressed as nM of MDA per mg of protein.163 The drawback of this assay is that TBA is not completely specific for MDA, and moreover the other types of compounds, such as, non-lipid-related materials as well as fatty peroxide-derived decomposition products also react with TBA.186
5.1.6.1. Assessment of chromosomal damage. In addition to identifying mutations of a particular gene, it is also important to analyze the presence of chromosomal aberrations, such as, chromosome breaks, fusions and abnormal segregation, and also the presence of micronuclei.
5.1.6.1.1. Chromosomal aberration analysis. This assay uses fluorescent in situ hybridization (FISH) to detect small deletions and duplications in the chromosomes which are not visible in microscopic analyses. FISH uses fluorescent probes such as small DNA strands that are complementary to specific parts of a chromosome and thus hybridize those, the hybridization could be visualized under fluorescence microscopy. Briefly, the methodology involves the incubation of cells with the nanoparticles for a pre-determined period and then allowed to grow in fresh medium for 24 h. Cells are then arrested in metaphase by the addition of colcemid solution, and then subjected to hypotonic treatment with warm 0.075 M KCl and subsequently fixed with fixative (3
:1 methanol:acetic acid solution). Metaphase spreads are prepared and FISH is then performed using telomere- and centromere-specific peptide nucleic acid (PNA) probes labeled with Cy(cyanine)-3 and FITC, and analyzed using the imaging system.187
5.1.6.1.2. Cytokinesis blocked micronucleus assay (CBMN). CBMN assay measures the chromosomal breakage occurring as a result of nanoparticles toxicity to the cells. Micronuclei are small nuclei formed from chromosomal fragments or whole chromosomes which failed to incorporate into one of the daughter cells after cell division. The CBMN assay measures the chromosomal breakage occurring as a result of nanoparticles toxicity, if any, to the cells. Formation of micronucleus could be examined under fluorescent microscope. Using FISH technique with specific probes targeted to centromere region, it is also possible to determine whether a fragment of chromosome (clastogenic event) or an entire chromosome (aneugenic effect) forms micronucleus. The major advantage of micronucleus assay is that it can detect both chromosomal and genetic mutations, whereas, the limitation of this assay is that it can be applied only to the dividing cells.172 Micronucleus formation can be visualized using the dye Hoechst 33258, a membrane permeable fluorescent DNA stain that intercalate in A–T rich regions of the DNA. Following incubation with the nanoparticles, the cells are washed with PBS and fixed overnight with fixative containing acetic acid–methanol (1
:3 v/v). Cells are then washed and fixed on slides and stained with Hoechst 33258 (10 μg ml−1). Slides are examined under fluorescent microscope to count the number of binucleated cells containing micronuclei and to calculate the fraction of cells containing micronuclei.
5.1.6.1.3. Identification of DNA base modifications: 8-hydroxyl-2′-deoxyguanosine (8-OHdG) detection using the enzyme linked immunosorbent assay (ELISA) technique. 8-OHdG, an indicator of oxidative damage of DNA, is a modified DNA base with a hydroxyl group at eighth position of guanine, and can be quantitatively detected by ELISA technique. The chemical reaction and methodology of this assay188 are schematized in Fig. 14 and 15, respectively.
 | ||
| Fig. 14 Structural representation of the conversion of 2′-deoxyguanosine to 8-hydroxyl-2′-deoxyguanosine (8-OHdG) and its detection using ELISA assay for the identification of DNA base modifications in nanoparticles-treated cells. | ||
5.1.6.1.4. Identification of gene mutations using Ames assay. This assay has been used to determine the mutagenicity of a variety of nanomaterials, by employing the bacteria lacking DNA repair mechanisms and requiring histidine for their growth (e.g. Salmonella typhimurium).189 Following exposure to the test sample whose genotoxicity is to be assessed, if the bacteria form colonies in histidine-free media, it indicates the reversion of bacterial phenotype to a histidine-positive phenotype due to a reverse mutation in the histidine locus. The methodology involves the incubation of overnight cultures of S. typhimurium strains with the test sample and mixing with sterile top agar containing 0.6% agar and 0.5% NaCl containing histidine–biotin, subsequently pouring onto minimal glucose agar plates and then incubating at 37 °C for 48 h. The revertant colonies are counted and the result is considered positive for genotoxicity if the count of reverting colonies comes out to be double compared to that of the negative control.190 However, this assay employs bacteria and not mammalian cells, and thus the results should be interpreted with caution. In any case, the Ames assay alone is not sufficient to assess genotoxicity, and thus other genotoxicity assays should be performed in addition to this assay.
Of various assays described above for the assessment of the genotoxicity, the most commonly used in vitro genotoxicity assays are the comet assay and the micronucleus assay.191
5.1.6.1.5. Assays for alterations in gene expression. Gene expression is a process by which the genetic information present in the gene is used in the synthesis of protein(s) required for cell growth and functioning. Gene expression profiling is a simultaneous measurement of the expression level of thousands of genes (particularly mRNA transcripts) to assess cell functioning. It discriminates between the cells that show active cell division or the cellular response following treatment with nanoparticles. Gene expression assays include Northern blot analysis, Ribonuclease protection assays (RPA), quantitative real-time polymerase chain reaction (qRT-PCR), and microarrays.132
5.1.6.1.5.1. Northern blot analysis. This method is considered as a powerful method to measure the levels of mRNA in a quantitative manner, and also provides information about the size and sequence of mRNA molecules. RNA isolation is performed from test sample-treated cells, separated on denaturing agarose gel containing 0.8 mol l−1 formaldehyde, and is then transferred onto nitrocellulose membrane. RNA on the membrane is hybridized to a radio-labeled RNA probe that is complementary to the target sequence. Membranes are then washed initially with 2× sodium chloride–sodium citrate solution and 0.1% SDS and then with 0.1× SSC and 0.1% SDS. The hybridized RNA-radiolabelled probe is then detected by autoradiograph analysis.192 The main drawback of this method is the need to use radioactive agents making the procedure time consuming and complicated to handle.
5.1.6.1.5.2. Ribonuclease protection assay. It is a sensitive method that simultaneously detects, quantifies and maps several RNA species. Following RNA isolation from nanoparticle treated cells, the target RNA sequence is hybridized with labelled antisense probes. Unhybridized probes and RNA sample are then removed by digestion with a mixture of nucleases. Nucleases are then inactivated, samples are digested with proteinase-K and extracted with phenol–chloroform mixture. Following ethanol precipitation with 4 M ammonium acetate solution, the hybridized samples are separated on Tris–borate–EDTA–urea gel, and subsequently analyzed using autoradiograph analysis.193 The main limitations of this assay are that this assay does not provide information about the transcript size, and the lack of probe flexibility.
5.1.6.1.5.3. Quantitative real-time polymerase chain reaction (qRT-PCR) or real time reverse transcription followed by polymerase chain reaction. It is one of the sensitive methods for the quantitative measurement of mRNA. This method uses dyes such as SYBR green (which emit fluorescence signal by binding to double stranded DNA), TaqMan (generates fluorescence depending on Forster resonance energy transfer-FRET by coupling of TaqMan and quencher to oligonucleotide substrates).194 Following total RNA isolation from test sample-treated cells, it is converted to single stranded cDNA (complementary DNA) in the presence of reverse transcriptase and oligo primers. Using this single stranded cDNA as a template, cDNA is amplified to double stranded DNA in the presence of specific primers and SYBR green dye.195 The fluorescence signal generated from the dye is proportional to the amount of double stranded DNA present in the sample, from which the level of expression of mRNA of the target gene can be determined.
5.1.6.1.5.4. Microarray. Microarray technology involves the hybridization of nucleic acid target sequence to a large set of oligonucleotide probes, for the determination of gene sequence or for the detection of variations in gene expression. Microarray technology aids in simultaneous examination of the expression of thousands of genes in a single RNA. In this technique, mRNA molecules from test sample-treated and untreated cells are isolated and converted to cDNA by RT-PCR using different fluorescent dyes. The resultant fluorescent labelled cDNAs are placed on microarray slide containing synthetic DNAs that are complementary to the fluorescent-labeled cDNAs, to result in hybridization (see also http://www.genome.gov/10000533). Bright fluorescence indicates overexpression of genes and lighter fluorescence indicates lower expression of genes. Thus, using this assay, the gene expression pattern, i.e., the types of genes upregulated and the types of genes downregulated in the cells due to nanoparticle-treatment can be analyzed. For instance, the incubation of mouse hepatic cells with poly(ethylene glycol)-block-polylactide nanoparticles, resulted in the over-expression of various ATP-binding cassette transporters and down-regulation of glutathione-S-transferase P1 as assessed using mouse cDNA microarray and validated by RT-PCR assays.196
5.2. Interaction with blood components
Nanoparticles, either injected parenterally into the body, or after exposure through the local routes (e.g. oral, inhalation, eye), come into contact with blood and thus with various blood components including blood cells and plasma proteins. Due to the unique physicochemical characteristics, the nanoparticles possess great potential to interact with various blood components, and such interaction needs to be well characterized and assessed from toxicological standpoint. Interaction of nanoparticles with blood components may be assessed for, (i) their compatibility with blood cells by the assessment of hemolytic potential and any alteration in hematological parameters, and (ii) potential to stimulate the immune system including the complement system, the blood coagulation and the inflammatory responses. Additionally, the assessment of nanoparticle–plasma protein interactions may also be considered to aid in nanoparticles toxicological assessment.Hemolysis is a damage caused to the red blood cells, leading to the leakage of hemoglobin into the blood. In vitro evaluation of the hemocompatibility of nanomaterials becomes an important part of in vitro characterization assays during their early preclinical development. Dobrovolskaia and colleagues197 at National Characterization Laboratory USA have validated in vitro assay protocol for the determination of hemolytic properties of the nanoparticles (also see http://ncl.cancer.gov/NCL_Method_ITA-1.pdf). In this assay, the nanoparticles are incubated with blood, and the undamaged RBC in the blood sample is separated by centrifugation. Hemoglobin released by the RBC following the damage caused by the nanoparticles is converted to methemoglobin and subsequently to cyanmethemoglobin in the presence of Drabkin's reagent (Drabkin's reagent is an aqueous solution of a mixture of potassium ferricyanide, sodium bicarbonate and potassium cyanide). The so formed cyanmethemoglobin is measured spectrophotometrically at 540 nm. The total hemoglobin in the whole blood is also quantified. Poly-L-lysine and Triton X-100 were suggested as positive controls, while PEG was suggested as negative control for hemolysis assessment.197 According to ASTM (American Society for Testing and Materials Designation, Standard practice for Assessment of Hemolytic Properties of Materials Designation F-756-00), <2% hemolysis is considered to be non-hemolytic; 2–5% is considered slightly hemolytic; >5% is considered hemolytic. Percent hemolysis of the samples is calculated using the formula below:

 | ||
| Fig. 15 Scheme showing the methodology of 8-hydroxyl-2′-deoxyguanosine detection for the identification of DNA base modifications using ELISA technique. | ||
In addition to the assessment of hemolytic potential, the hemocompatibility of the nanoparticles could be assessed by performing whole blood cell count including erythrocytes, leucocytes and thrombocytes (platelets), and verifying the differences, if any, in the blood cell count in the nanoparticles-treated blood samples in comparison to that of the untreated blood samples.
The thrombogenicity of nanoparticles may be assessed by measuring their potential to induce platelet aggregation and modify the coagulation time.203 Platelet aggregation assay involves the incubation of nanoparticles sample with platelet rich plasma (PRP) obtained from the freshly derived whole human blood. After 15 min of incubation, the PRP is analyzed using particle count and size analyser to determine the number of active platelets. The percent platelet count is calculated using the formula below. The percent platelet aggregation above 20% indicates that the nanoparticles are thrombogenic.

The plasma coagulation time can be assessed by exposing the platelet-poor plasma from whole human blood, followed by analysis of prothrombin, activated partial thromboplastin time and thrombin time.204,205 Briefly, one volume of nanoparticles as aqueous suspension at defined concentrations is incubated with nine volumes of platelet-poor plasma at 37 °C, followed by the assessment of coagulation using coagulometer.204 Additionally, the effects of nanoparticles on plasma clotting factors can be assessed by diluting the test nanoparticle suspension in plasma which is deficient in the clotting factor the impact on which has to be measured, and then comparing the clotting time with that of normal plasma.204 For instance, using this assay, the effect of non-PEGylated poly(lactic acid) and PEGylated poly(lactic acid) nanoparticles on plasma clotting time and on clotting factors has been assessed.204 Interestingly, non-PEGylated nanoparticles prolonged considerably the clotting time and have also reduced slightly the levels of factors VIII, IX, VII and X but decreased factor V level by 20–30%. On the opposite PEGylated nanoparticles did not show any effect on coagulation time and on clotting factors,204 thus suggesting the importance of PEG in minimizing the interaction of nanoparticles with coagulation system.
Methodologically,38 the plasma protein corona on nanoparticles can be analyzed by incubating the nanoparticles samples with plasma in 10 mM phosphate, 0.15 M NaCl, 1 mM EDTA for 1 h. The samples are centrifuged to recover the nanoparticle–protein complexes. Proteins are eluted from nanoparticles by adding SDS-sample buffer, and the eluted proteins are then separated on 12% SDS-PAGE one dimensional gel. Bands of interest from the gels are eluted and digested with trypsin and the resultant peptide mixtures are subsequently resuspended in 0.1% formic acid and analyzed using liquid chromatography mass spectrometry for the determination of the qualitative and quantitative composition of the protein corona. These proteins are classified based on their physicochemical and biological properties using relevant bioinformatic tools.211 Alternatively, techniques such as Protein Lab-on-Chip® and capillary electrophoresis have been also reported for the assessment of serum protein adsorption onto nanoparticles surfaces.212 The Protein Lab-on-Chip® allowed the determination of kinetics of protein adsorption onto the nanoparticles surfaces.
6. Nanoparticles toxicity assessment: in vivo
6.1. Key considerations
In vivo assessment of the toxicity of nanoparticles is crucial to understand their behaviour in the complex biological environment and to anticipate their safety in the intended species, animal or human. Additionally, the information on the metabolization potential of nanoparticles in the extracellular environment, the environment which the nanoparticles encounter before coming into contact with the cells, could be possible with in vivo assays but may be very challenging with in vitro assays. The in vivo assessment should include appropriate assays to understand the pharmacokinetics and biodistribution of nanoparticles, and their effects at the blood and the tissue level in short-term and long-term after administration of single and multiple doses relevant to the intended route of administration and application. Such assays are expected to provide information on the dose-dependent accumulation in desired and in undesired tissues, the concentration within the tissues, and the residence time in the tissues. Based on the above information, one can go back to the bench to understand in vitro the mechanism of degradation and/or toxicity of nanoparticles in corresponding cell type and at corresponding concentrations to assess the acceptability level of toxicity and to anticipate the tolerable doses in vivo. Additionally, assessment of kinetics of degradation/metabolism of nanoparticles in the body allows defining the frequency of administration of nanoparticles product to the patients.The biodistribution studies become further important when nanoparticles are associated with a therapeutic agent, for instance for drug delivery purposes. The safety of nanoparticle-associated therapeutic agent mainly depends on its affinity to the nanoparticle and on the nanoparticle biodistribution. If the nanoparticles distribute to the undesired compartments in the body and reside in these compartments for longer time, then safety is a measure of not only the effect of nanoparticle carrier but also of the effects caused by therapeutic agent, especially if the agent is cytotoxic or its target lies in these compartments. For instance, in case of polyethylene glycol-coated liposomal doxorubicin, one of the safety concerns in the patients treated with this product is the hand-foot syndrome (also called as ‘palmar-plantar erythrodysesthesia’)213 resulting from extravasation of the drug-containing small liposomes into skin layers and subsequent release of the cytotoxic drug doxorubicin in the local tissue, leading to tissue irritation and inflammation.214 Thus, the rate of clearance or degradation kinetics of nanoparticles in the accumulated tissues determines the local safety of nanoparticles in these tissues.215 In addition, the metabolites emerging from nanoparticle degradation, in case toxic, may also be responsible for inducing local tissue toxicity. Thus, the in vivo assays of toxicity assessment should include all possible means of understanding the nanoparticles fate and its impact on the complex biological system, to de-risk the nanoparticle product development toward the intended use.
Attention is also needed with respect to the interpretation of pharmacokinetic/biodistribution data. It is known that prolonged systemic circulation of the nanoparticles could be achieved by coating their surface with hydrophilic polymers such as polyethylene glycol due to its ability to mask the nanoparticle surface from recognition by the body's immune system and consequently limit the nanoparticles uptake by macrophages. Alternatively, the nanoparticles not coated with PEG may also exhibit prolonged systemic circulation at doses exceeding the phagocytic capacity of the macrophages, owing to the macrophage saturation. This may induce complications related to the blockade of macrophages' essential functions including increased risk of infections in the body.216 Similarly, in case of macrophage saturation effect, the nanoparticles may not exhibit linear pharmacokinetics with dose, because of the depletion of macrophage uptake capacity at increasing nanoparticles doses. Such effect may have implications on the predictability of the dose response and extrapolation of pharmacokinetic data. Thus, understanding of the macrophage saturation potential and in turn the recovery of macrophage activity in vivo, following systemic exposure of the nanoparticles, is critical to anticipate the potential hazards, and also to allow designing of injection schedule with time spacing between the two doses appropriate to avoid macrophage saturation and the associated undesired effects.
With nanoparticles-based products, two types of variability in performances may emerge, such as, the variability linked to the nanoparticles physicochemical attributes, and the variability linked to the patient pathophysiology. While it is crucial to ensure reproducibility of the physicochemical attributes of nanoparticles formulations from chemistry, manufacturing and controls (CMC) standpoint in all manufacturing lots, taking into account the potential variability associated with patient pathophysiology is of equal importance as early as in preclinical development by the design of relevant animal models for the proof of concept evaluation. For instance, the distribution of nanoparticles to healthy tissues may be considerably different from their distribution to the same tissues but in a pathological state, such as, inflammatory condition, compromised tissue endothelial wall, etc. Moreover, the variations in other tissue-related parameters such as enzymatic composition, pH, etc. may have impact on the nanoparticle degradation kinetic and the release of encapsulated payload from the nanoparticles. Thus, a prudent choice of in vivo models having pathophysiology closer to that of the target species for which the product is to be developed, and the target indication in these species. Moreover, development of appropriate biopharmaceutical modeling approaches may allow better predictability of nanoparticle product performances and increase the probability of translation of preclinical proof-of-concept to clinic.
6.2. Assays
7. Physiological based modeling of nanoparticles
Physiological based modeling is extensively used in pharmaceutical development, for the prediction of pharmacokinetic parameters of drug formulations in humans from data obtained in preclinical models and across various preclinical species, by the integration of anatomical, physiological and biochemical aspects of the body and segmenting various organs into individual compartments and interconnecting those with the mass transportation and ADME properties of drugs.228 Application of the classical physiology based modeling, generally used for drug development, to the nanoparticles may not be straightforward because of the complexity associated with the interplay between nanoparticles' physicochemical characteristics and biophysical properties, biodistribution, metabolism, etc. Design of modeling approaches suitable for nanoparticles could be built on existing classical modeling experience, by taking into account additional nanoparticle-specific factors, such as, clearance by macrophages of RES organs and other mechanisms, differential extravasation through vascular endothelia of varying fenestrae dimensions, distribution influenced by plasma protein adsorption, cellular uptake and the associated kinetics and mechanisms, biodegradation, and elimination kinetics, etc.For instance, MacCalman and coworkers229 reported a model to describe the body biodistribution of nanoparticles after administration by inhalation to rat. In this report, the distribution of inhaled nanoparticles from lungs to major organs of the body, such as, liver, kidney, spleen, heart, brain and gastrointestinal tract was modeled. Various regions within lung were considered as different compartments, and the translocation of particles to blood vessels and their subsequent distribution to other organs were integrated into the model. Noteworthy, the model fit was found good for the total particle mass measured in the body and to the particle mass measured in some organs, such as, lung, brain, spleen but not for organs such as liver and kidney. Moreover, the optimal parameter estimates for model were suggested to be dependent on nanoparticles' physicochemical characteristics and also on the routes of nanoparticles exposure to lung. Similarly, a model for describing the pulmonary retention and clearance of inhaled silica crystals in rat (median diameters of the order of 1.7 μm) and responses to dose, such as, inflammatory response and fibrosis has been reported.230 Some studies have also reported attempts to build models to define relationship between the properties of nanoparticles and their biodistribution,231–233 wherein model with appropriate fit simulating the experimental biodistribution data of nanoparticle formulations with varying characteristics has been described by Li and coworkers.233 On the other hand, a model describing the simulation of time-dependent tissue distribution kinetics of quantum dot nanoparticles up to a period of 6 months after intravenous injection to mice has been also reported,234 wherein the prediction of tissue distribution kinetics by this model was found to be consistent with the experimental data.
Overall, designing of relevant modeling approaches for the prediction of performance and potential toxicities of new nanomaterials based on their material properties and physicochemical characteristics, although challenging, could be of high value for the design and development of nanoparticles exhibiting desired biopharmaceutical performance and safety, and also in regulatory perspective.
8. Conclusion
Nanoparticles, owing to their unique physicochemical characteristics, possess great potential to interact with the biological components, such as, blood, plasma proteins, tissues, cells and sub-cellular components. The nanoparticles designed from new materials with varying architectures and functionalities aimed at enhancing the performance may potentially exhibit varying physicochemical characteristics and thus may have varying impact on the nanoparticles' biopharmaceutical properties. Thus, delineating the interplay between the physicochemistry and biophysical properties of nanoparticles, and their impact on the pharmacokinetic (absorption, distribution, metabolism and excretion) and toxicological properties, could be expected to help in designing of appropriate nanoparticles products for biomedical applications to human or veterinary use. A thorough in vitro toxicological assessment with mechanistic understanding is considered useful in anticipating the potential in vivo toxicity, and hence is expected to be valuable for extrapolation of preclinical data to human. The development of physiological-based computational models allowing prediction of ADME properties and toxicity, from the material and physicochemical characteristics may help in anticipating potential safety issues, and thus likely pave way toward the successful design of nanoparticles with desired biopharmaceutical performance and safety.Disclaimer
The views expressed by the authors in this review do not necessarily reflect the opinions of Sanofi.References
- D. V. Bazile, J. Drug Delivery Sci. Technol., 2014, 24, 12 CrossRef CAS.
- M. Johannsen, U. Gneveckow, B. Thiesen, K. Taymoorian, C. H. Cho, N. Waldöfner, R. Scholz, A. Jordan, S. A. Loening and P. Wust, Eur. Urol., 2007, 52, 1653 CrossRef PubMed.
- M. Johannsen, B. Thiesen, P. Wust and A. Jordan, Int. J. Hyperthermia, 2010, 26, 790 CrossRef PubMed.
- E. A. Neuwelt, P. Várallyay, A. G. Bagó, L. L. Muldoon, G. Nesbit and R. Nixon, Neuropathol. Appl. Neurobiol., 2004, 30, 456 CrossRef CAS PubMed.
- S. R. Digumarthy, D. V. Sahani and S. Saini, Canc. Imag., 2005, 5, 20 CrossRef PubMed.
- L. H. Reddy, J. L. Arias, J. Nicolas and P. Couvreur, Chem. Rev., 2012, 112, 5818 CrossRef CAS PubMed.
- K. K. L. Phua, H. F. Staats, K. W. Leong and S. K. Nair, Sci. Rep., 2014, 4, 5128 CAS.
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971 RSC.
- M. Johannsen, B. Thiesen, P. Wust and A. Jordan, Int. J. Hyperthermia, 2010, 26, 790 CrossRef PubMed.
- H. S. Huang and J. F. Hainfeld, Int. J. Nanomed., 2013, 8, 2521 Search PubMed.
- F. Liu, S. Laurent, H. Fattahi, L. Vander Elst and R. N. Muller, Nanomedicine, 2011, 6, 519 CrossRef CAS PubMed.
- J. L. Arias, L. H. Reddy, M. Othman, B. Gillet, D. Desmaële, F. Zouhiri, F. Dosio, R. Gref and P. Couvreur, ACS Nano, 2011, 5, 1513 CrossRef CAS PubMed.
- J. Xie, S. Lee and X. Chen, Adv. Drug Delivery Rev., 2010, 62, 1064 CrossRef CAS PubMed.
- T. Dvir, B. P. Timko, D. S. Kohane and R. Langer, Nat. Nanotechnol., 2011, 6, 13 CrossRef CAS PubMed.
- L. M. Graham, T. M. Nguyen and S. B. Lee, Nanomedicine, 2011, 6, 759 CrossRef PubMed.
- Y. Hoshino, H. Koide, K. Furuya, W. W. Haberaecker III, S. H. Lee, T. Kodama, H. Kanazawa, N. Oku and K. J. Shea, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 33 CrossRef CAS PubMed.
- J. A. H. Junghanns and R. H. Müller, Int. J. Nanomed., 2008, 3, 295 CAS.
- B. E. Rabinow, Nat. Rev. Drug Discovery, 2004, 3, 785 CrossRef CAS PubMed.
- W. T. Lim, E. H. Tan, C. K. Toh, S. W. Hee, S. S. Leong, P. C. S. Ang, N. S. Wong and B. Chowbay, Ann. Oncol., 2010, 21, 382 CrossRef CAS PubMed.
- E. Miele, G. P. Spinelli, E. Miele, F. Tomao and S. Tomao, Int. J. Nanomed., 2009, 4, 99 CrossRef CAS.
- A. Gabizon, H. Shmeeda and Y. Barenholz, Clin. Pharmacokinet., 2003, 42, 419 CrossRef CAS PubMed.
- J. Hrkach, D. Von Hoff, M. Mukkaram Ali, E. Andrianova, J. Auer, T. Campbell, D. De Witt, M. Figa, M. Figueiredo, A. Horhota, S. Low, K. McDonnell, E. Peeke, B. Retnarajan, A. Sabnis, E. Schnipper, J. J. Song, Y. H. Song, J. Summa, D. Tompsett, G. Troiano, T. Van Geen Hoven, J. Wright, P. LoRusso, P. W. Kantoff, N. H. Bander, C. Sweeney, O. C. Farokhzad, R. Langer and S. Zale, Sci. Transl. Med., 2012, 4, 128ra39 Search PubMed.
- C. Mamot, R. Ritschard, A. Wicki, G. Stehle, T. Dieterle, L. Bubendorf, C. Hilker, S. Deuster, R. Herrmann and C. Rochlitz, Lancet Oncol., 2012, 13, 1234 CrossRef CAS.
- P. J. Gaillard, B. M. Kerklaan, P. Aftimos, S. Altintas, A. Jager, W. Gladdines, F. Lonnqvist, P. Soetekouw, H. Verheul, A. Awada, J. Schellens and D. Brandsma, Cancer Res., 2014, 74, CT216 Search PubMed.
- M. K. Yu, J. Park and S. Jon, Theranostics, 2012, 2, 3 CrossRef CAS PubMed.
- T. Coelho, D. Adams, A. Silva, P. Lozeron, P. N. Hawkins, T. Mant, J. Perez, J. Chiesa, S. Warrington, E. Tranter, M. Munisamy, R. Falzone, J. Harrop, J. Cehelsky, B. R. Bettencourt, M. Geissler, J. S. Butler, A. Sehgal, R. E. Meyers, Q. Chen, T. Borland, R. M. Hutabarat, V. A. Clausen, R. Alvarez, K. Fitzgerald, C. Gamba-Vitalo, S. V. Nochur, A. K. Vaishnaw, D. W. Sah, J. A. Gollob and O. B. Suhr, N. Engl. J. Med., 2013, 369, 819 CrossRef CAS PubMed.
- M. E. Davis, J. E. Zuckerman, C. H. Choi, D. Seligson, A. Tolcher, C. A. Alabi, Y. Yen, J. D. Heidel and A. Ribas, Nature, 2010, 464, 1067 CrossRef CAS PubMed.
- C. Butts, A. Maksymiuk, G. Goss, D. Soulières, E. Marshall, Y. Cormier, P. M. Ellis, A. Price, R. Sawhney, F. Beier, M. Falk and N. Murray, J. Cancer Res. Clin. Oncol., 2011, 137, 1337 CrossRef CAS PubMed.
- P. O. Ilyinskii, C. J. Roy, C. P. O'Neil, E. A. Browning, L. A. Pittet, D. H. Altreuter, F. Alexis, E. Tonti, J. Shi, P. A. Basto, M. Iannacone, A. F. Radovic-Moreno, R. S. Langer, O. C. Farokhzad, U. H. von Andrian, L. P. Johnston and T. K. Kishimoto, Vaccine, 2014, 32, 2882 CrossRef CAS PubMed.
- K. K. Jain, Drug Discovery Today, 2005, 10, 1435 CrossRef CAS.
- E. C. Cho, C. Glaus, J. Chen, M. J. Welch and Y. Xia, Trends Mol. Med., 2010, 16, 561 CrossRef CAS PubMed.
- M. A. Hahn, A. K. Singh, P. Sharma, S. C. Brown and B. M. Moudgil, Anal. Bioanal. Chem., 2011, 399, 3 CrossRef CAS PubMed.
- J. Liu, M. Yu, C. Zhou and J. Zheng, Mater. Today, 2013, 16, 477 CrossRef CAS PubMed.
- J. Liu, M. Yu, X. Ning, C. Zhou, S. Yang and J. Zheng, Angew. Chem., Int. Ed., 2013, 52, 12572 CrossRef CAS PubMed.
- J. Liu, M. Yu, C. Zhou, S. Yang, X. Ning and J. Zheng, J. Am. Chem. Soc., 2013, 135, 4978 CrossRef CAS PubMed.
- S. Mura and P. Couvreur, Adv. Drug Delivery Rev., 2012, 64, 1394 CrossRef CAS PubMed.
- T. H. Kim, S. Lee and X. Chen, Expert Rev. Mol. Diagn., 2013, 13, 257 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265 CrossRef CAS PubMed.
- S. Milani, F. B. Bombelli, A. S. Pitek, K. A. Dawson and J. Rädler, ACS Nano, 2012, 6, 2532 CrossRef CAS PubMed.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543 CrossRef CAS PubMed.
- S. R. Saptarshi, A. Duschl and A. L. Lopata, J. Nanobiotechnol., 2013, 11, 26 CrossRef CAS PubMed.
- N. Wangoo, C. R. Suri and G. Shekhawat, Appl. Phys. Lett., 2008, 92, 133104 CrossRef PubMed.
- Z. N. Gheshlaghi, G. H. Riazi, S. Ahmadian, M. Ghafari and R. Mahinpou, Acta Biochim. Biophys. Sin., 2008, 40, 777 CrossRef CAS PubMed.
- M. Mahmoudi, M. A. Shokrgozar, S. Sardari, M. K. Moghadam, H. Vali, S. Laurent and P. Stroeve, Nanoscale, 2011, 3, 1127 RSC.
- L. Treuel, S. Brandholt, P. Maffre, S. Wiegele, L. Shang and G. U. Nienhaus, ACS Nano, 2014, 8, 503 CrossRef CAS PubMed.
- A. Gessner, A. Lieske, B. Paulke and R. Müller, Eur. J. Pharm. Biopharm., 2002, 54, 165 CrossRef CAS.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. Chan, J. Am. Chem. Soc., 2012, 134, 2139 CrossRef CAS PubMed.
- C. Fang, B. Shi, Y. Y. Pei, M. H. Hong, J. Wu and H. Z. Chen, Eur. J. Pharm. Sci., 2006, 27, 27 CrossRef CAS PubMed.
- P. Varallyay, G. Nesbit, L. L. Muldoon, R. R. Nixon, J. Delashaw, J. I. Cohen, A. Petrillo, D. Rink and E. A. Neuwelt, Am. J. Neuroradiol., 2002, 23, 510 Search PubMed.
- C. Chouly, D. Pouliquen, I. Lucet, J. J. Jeune and P. J. Jallet, J. Microencapsulation, 1996, 13, 245 CrossRef CAS PubMed.
- Z. J. Deng, M. Liang, I. Toth, M. Monteiro and R. F. Minchin, Nanotoxicology, 2013, 7, 314 CrossRef CAS PubMed.
- A. Gessner, A. Lieske, B. Paulke and R. Muller, J. Biomed. Mater. Res., Part A, 2003, 65, 319 Search PubMed.
- A. A. Vertegel, R. W. Siegel and J. S. Dordick, Langmuir, 2004, 20, 6800 CrossRef CAS PubMed.
- Z. Deng, G. Mortimer, T. Schiller, A. Musumeci, D. Martin and R. Minchin, Nanotechnology, 2009, 20, 455101 CrossRef PubMed.
- M. S. Ehrenberg, A. E. Friedman, J. N. Finkelstein, G. Oberdorster and J. L. McGrath, Biomaterials, 2009, 30, 603 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, P. Aggarwal, J. B. Hall and S. E. McNeil, Mol. Pharm., 2008, 5, 487 CAS.
- D. E. Owens III and N. A. Peppas, Int. J. Pharm., 2006, 307, 93 CrossRef PubMed.
- T. Ishida, H. Harashima and H. Kiwada, Curr. Drug Metab., 2001, 2, 397 CrossRef CAS.
- J. Kreuter, J. Nanosci. Nanotechnol., 2004, 4, 484 CrossRef CAS PubMed.
- K. Ogawara, K. Furumoto, S. Nagayama, K. Minato, K. Higaki, T. Kai and T. Kimura, J. Controlled Release, 2004, 100, 451 CrossRef CAS PubMed.
- A. Akinc, W. Querbes, S. De, J. Qin, M. Frank-Kamenetsky, K. N. Jayaprakash, M. Jayaraman, K. G. Rajeev, W. L. Cantley, J. R. Dorkin, J. S. Butler, L. Qin, T. Racie, A. Sprague, E. Fava, A. Zeigerer, M. J. Hope, M. Zerial, D. W. Sah, K. Fitzgerald, M. A. Tracy, M. Manoharan, V. Koteliansky, A. de Fougerolles and M. A. Maier, Mol. Ther., 2010, 18, 1357 CrossRef CAS PubMed.
- J. R. Dunkelberger and W. C. Song, Cell Res., 2010, 20, 34 CrossRef CAS PubMed.
- J. Szebeni, Toxicology, 2005, 216, 106 CrossRef CAS PubMed.
- S. M. Moghimi and Z. S. Farhangrazi, Nanomedicine: Nanotechnology, Biology and Medicine, 2013, 9, 458 CrossRef CAS PubMed.
- I. Bertholon, C. Vauthier and D. Labarre, Pharm. Res., 2006, 23, 1313 CAS.
- J. Szebeni, P. Bedocs, Z. Rozsnyay, Z. Weiszhár, R. Urbanics, L. Rosivall, R. Cohen, O. Garbuzenko, G. Báthori, M. Tóth, R. Bünger and Y. Barenholz, Nanomedicine, 2012, 8, 176 CAS.
- C. Salvador-Morales, L. Zhang, R. Langer and O. C. Farokhzad, Biomaterials, 2009, 30, 2231 CrossRef CAS PubMed.
- M. M. Markiewski, B. Nilsson, K. N. Ekdahl, T. E. Mollnes and J. D. Lambris, Trends Immunol., 2007, 28, 184 CrossRef CAS PubMed.
- M. M. Markiewski, R. A. DeAngelis and J. D. Lambris, Mol. Immunol., 2006, 43, 45 CrossRef CAS PubMed.
- D. Mastellos, C. Andronis, A. Persidis and J. D. Lambris, Clin. Immunol., 2005, 115, 225 CrossRef CAS PubMed.
- P. Tan, F. W. Luscinskas and S. Homer-Vanniasinkam, Eur. J. Vasc. Endovasc. Surg., 1999, 17, 373 CrossRef CAS PubMed.
- M. A. Dobrovolskaia and S. E. McNeil, Nat. Nanotechnol., 2007, 2, 469 CrossRef CAS PubMed.
- M. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol. Rev., 2001, 53, 283 Search PubMed.
- L. H. Reddy and P. Couvreur, J. Hepatol., 2011, 55, 1461 CrossRef CAS PubMed.
- S. W. Jones, R. A. Roberts, G. R. Robbins, J. L. Perry, M. P. Kai, K. Chen, T. Bo, M. E. Napier, J. P. Y. Ting, J. M. DeSimone and J. E. Bear, J. Clin. Invest., 2013, 123, 3061 CAS.
- C. D. Mills, K. Kincaid, J. M. Alt, M. J. Heilman and A. M. Hill, J. Immunol., 2000, 164, 6166 CrossRef CAS.
- D. V. Bazile, M. T. Bassoulet, M. Marlard, G. Splenlehauer and M. Veillard, J. Pharm. Sci. Technol., Jpn., 1993,(suppl. 53), 10 CAS.
- R. Gref, Y. Minamitake, M. T. Peracchia, V. Trubetskoy, V. Torchilin and R. Langer, Science, 1994, 263, 1600 CAS.
- S. I. Jeon, J. H. Lee, J. D. Andrade and P. G. De Gennes, J. Colloid Interface Sci., 1991, 142, 149 CrossRef CAS.
- R. Gref, M. Luck, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. H. Muller, Colloids Surf., B, 2000, 18, 301 CrossRef CAS.
- D. Bazile, C. Prud'homme, M. T. Bassoullet, M. Marlard, G. Spenlehauer and M. Veillard, J. Pharm. Sci., 1995, 84, 493 CrossRef CAS PubMed.
- P. Laverman, et al., Factors affecting the accelerated blood clearance of polyethylene glycol-liposomes upon repeated injection, J. Pharmacol. Exp. Ther., 2001, 298, 607–612 CAS.
- R. Saadati, S. Dadashzadeh, Z. Abbasian and H. Soleimanjahi, Pharm. Res., 2013, 30, 985 CAS.
- T. L. Cheng, P. Y. Wu, M. F. Wu, J. W. Chern and S. R. Roffler, Bioconjugate Chem., 1999, 10, 520 CrossRef CAS PubMed.
- A. W. Richter and E. Akerblom, Int. Arch. Allergy Appl. Immunol., 1984, 74, 36 CAS.
- J. K. Armstrong, G. Hempel, S. Koling, L. S. Chan, T. Fisher, H. J. Meiselman and G. Garratty, Cancer, 2007, 110, 103 CrossRef PubMed.
- H. Schellekens, W. E. Hennink and V. Brinks, Pharm. Res., 2013, 30, 1729 CrossRef CAS PubMed.
- T. Ishida, X. Wang, T. Shimizu, K. Nawata and H. Kiwada, J. Controlled Release, 2007, 122, 349 CrossRef CAS PubMed.
- X. Wang, T. Ishida and H. Kiwada, J. Controlled Release, 2007, 119, 236 CrossRef CAS PubMed.
- M. R. Sherman, L. D. Williams, M. A. Sobczyk, S. J. Michaels and M. G. P. Saifer, Role of the Methoxy Group in Immune Responses to mPEG-Protein Conjugates, Bioconjugate Chem., 2012, 23, 485–499 CrossRef CAS PubMed.
- T. Ishihara, M. Takeda, H. Sakamoto, A. Kimoto, C. Kobayashi, N. Takasaki, K. Yuki, K. Tanaka, M. Takenaga, R. Igarashi, T. Maed, N. Yamakawa, Y. Okamoto, M. Otsuka, T. Ishida, H. Kiwada, Y. Mizushima and T. Mizushima, Pharm. Res., 2009, 26, 2270 CAS.
- F. Gentile, M. Ferrari and P. Decuzzi, Ann. Biomed. Eng., 2008, 36, 254 CrossRef PubMed.
- T. Horn, J. H. Henriksen and P. Christoffersen, Liver Int., 1986, 6, 98 CAS.
- E. Igarashi, Factors affecting toxicity and efficacy of polymeric nanomedicines, Toxicol. Appl. Pharmacol., 2008, 229, 121 CrossRef CAS PubMed.
- S. M. Moghimi and I. Hamad, Factors controlling pharmacokinetics of intravenously injected nanoparticulate systems, in Nanotechnology in drug delivery Biotechnology: Pharmaceutical aspects, ed. M. M. de Villiers, P. Aramwit and G. S. Kwon, Springer, New York, 2009, vol. X, pp. 267–282 Search PubMed.
- H. Sarin, J. Angiog. Res., 2010, 2, 14 Search PubMed.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165 CrossRef CAS PubMed.
- C. H. J. Choi, J. E. Zuckerman, P. Webster and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6656 CrossRef CAS PubMed.
- T. Fujita, M. Nishikawa, Y. Ohtsubo, J. Ohno, Y. Takakura, H. Sezaki and M. Hashida, J. Drug Targeting, 1994, 2, 157 CrossRef CAS PubMed.
- S. D. Li and L. Huang, Mol. Pharm., 2008, 5, 496 CrossRef CAS PubMed.
- T. S. Levchenko, R. Rammohan, A. N. Lukyanov, K. R. Whiteman and V. P. Torchilin, Int. J. Pharm., 2002, 240, 95 CrossRef CAS.
- R. Kedmi, N. Ben-Arie and D. Peer, Biomaterials, 2010, 31, 6867 CrossRef CAS PubMed.
- J. A. Champion and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4930 CrossRef CAS PubMed.
- J. A. Champion and S. Mitragotri, Pharm. Res., 2009, 26, 244 CrossRef CAS PubMed.
- S. Tollis, A. E. Dart, G. Tzircotis and R. G. Endres, BMC Syst. Biol., 2010, 4, 149 CrossRef PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249 CrossRef CAS PubMed.
- A. Maksimenko, F. Dosio, J. Mougin, A. Ferrero, S. Wack, L. H. Reddy, A. A. Weyn, E. Lepeltier, C. Bourgaux, B. Stella, L. Cattel and P. Couvreur, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E217 CrossRef CAS PubMed.
- Z. Chu, S. Zhang, B. Zhang, C. Zhang, C. Y. Fang, I. Rehor, P. Cigler, H. C. Chang, G. Lin, R. Liu and Q. Li, Sci. Rep., 2014, 4, 4495 Search PubMed.
- R. E. Yanes, D. Tarn, A. A. Hwang, D. P. Ferris, S. P. Sherman, C. R. Thomas, J. Lu, A. D. Pyle, J. I. Zink and F. Tamanoi, Small, 2013, 9, 697 CrossRef CAS PubMed.
- K. C. L. Black, Y. Wang, H. P. Luehmann, X. Cai, W. Xing, B. Pang, Y. Zhao, C. S. Cutler, L. V. Wang, Y. Liu and Y. Xia, ACS Nano, 2014, 8, 4385 CrossRef CAS PubMed.
- K. A. Beningo and Y. L. Wang, Fc-receptor-mediated phagocytosis is regulated by mechanical properties of the target, J. Cell Sci., 2002, 115, 849 CAS.
- K. Shimizu, Y. Maitani, K. Takayama and T. Nagai, J. Drug Targeting, 1996, 4, 245 CrossRef CAS PubMed.
- E. Igarashi, in Nanomedicines and nanoproducts – Applications, disposition and toxicology in human body, ed. E. Igarashi, CRC press, Boca Raton, Florida, 2015, pp. 165–216 Search PubMed.
- K. Muramatsu, Y. Maitani, K. Takayama and T. Nagai, Drug Dev. Ind. Pharm., 1999, 25, 1099 CrossRef CAS PubMed.
- K. Muramatsu, T. Masumizu, Y. Maitani, S. H. Hwang, M. Kohno, K. Takayama and T. Nagai, Chem. Pharm. Bull., 2000, 48, 610 CrossRef CAS.
- S. M. Moghimi and H. M. Patel, FEBS Lett., 1988, 233, 143 CrossRef CAS.
- S. C. Semple, S. K. Klimuk, T. O. Harasym, N. Dos Santos, S. M. Ansell, K. F. Wong, N. Maurer, H. Stark, P. R. Cullis, M. J. Hope and P. Scherrer, Biochim. Biophys. Acta, 2001, 1510, 152 CAS.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323 CrossRef CAS PubMed.
- H. M. Burt, X. Zhang, P. Toleikis, L. Embree and W. L. Hunter, Colloids Surf., B, 1999, 16, 161 CrossRef CAS.
- S. K. Sahoo, J. Panyam, S. Prabha and V. Labhasetwar, J. Controlled Release, 2002, 82, 105 CrossRef CAS.
- L. Horev-Azaria, C. J. Kirkpatrick, R. Korenstein, P. N. Marche, O. Maimon, J. Ponti, R. Romano, F. Rossi, U. Golla-Schindler, D. Sommer, C. Uboldi, R. E. Unger and C. Villiers, Toxicol. Sci., 2011, 122, 489 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS.
- J. C. Stockert, A. Blazquez-Castro, M. Canete, R. W. Horobin and A. Villanueva, Acta Histochem., 2012, 114, 785 CrossRef CAS PubMed.
- N. J. Marshall, C. J. Goodwin and S. J. Holt, Growth Regul., 1995, 5, 69 CAS.
- M. Natarajan, S. Mohan, B. R. Martinez, M. L. Meltz and T. S. Herman, Cancer Detect. Prev., 2000, 24, 405 CAS.
- P. Takhar and S. Mahant, Arch. Appl. Sci. Res., 2011, 3, 389 CAS.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Eur. J. Biochem., 2000, 267, 5421 CrossRef.
- L. Horev-Azaria, G. Baldi, D. Beno, D. Bonacchi, U. Golla-Schindler, J. C. Kirkpatrick, S. Kolle, R. Landsiedel, O. Maimon, P. N. Marche, J. Ponti, R. Romano, F. Rossi, D. Sommer, C. Uboldi, R. E. Unger, C. Villiers and R. Korenstein, Part. Fibre Toxicol., 2013, 10, 32 CrossRef CAS PubMed.
- M. V. Lancaster and R. D. Fields, U.S. Pat., 5 501 959, 1996.
- S. M. Hussain and J. M. Frazier, Toxicol. Sci., 2002, 69, 424 CrossRef CAS PubMed.
- V. W. Hu, G. E. Black, A. Torres-Duarte and F. P. Abramson, FASEB J., 2002, 16, 1456 CAS.
- J. M. Hillegass, A. Shukla, S. A. Lathrop, M. B. MacPherson, N. K. Fukagawa and B. T. Mossman, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2, 219 CAS.
- E. Herzog, A. Casey, F. M. Lyng, G. Chambers, H. J. Byrne and M. Davoren, Toxicol. Lett., 2007, 174, 49 CrossRef CAS PubMed.
- M. Huang, E. Khor and L. Y. Lim, Pharmacol. Res., 2004, 21, 344 CAS.
- K. Kostarelos, L. Lacerda, G. Pastorin, W. Wu, S. Wieckowski, J. Luangsivilay, S. Godefroy, D. Pantarotto, J. P. Briand, S. Muller, M. Prato and A. Bianco, Nat. Nanotechnol., 2007, 2, 108 CrossRef CAS PubMed.
- W. F. Vevers and A. N. Jha, Ecotoxicology, 2008, 17, 410 CrossRef CAS PubMed.
- V. Sharma, D. Anderson and A. Dhawan, Apoptosis, 2012, 17, 852 CrossRef CAS PubMed.
- W. Strober, Curr. Protoc. Immunol., 1997, 21, A.3B.1 Search PubMed.
- V. Katsares, A. Petsa, A. Felesakis, Z. Paparidis, E. Nikolaidou, S. Gargani, I. Karvounidou, K. A. Ardelean, N. Grigoriadis and J. Grigoriadis, Lab. Med., 2009, 40, 557 CrossRef.
- S. Kanagesan, M. Hashim, S. Tamilselvan, N. B. Alitheen, I. Ismail and G. Bahmanrokh, J. Nanomater., 2013, 2013, 865024 Search PubMed.
- L. H. Reddy, J. M. Renoir, V. Marsaud, S. Lepetre-Mouelhi, D. Desmaële and P. Couvreur, Mol. Pharm., 2009, 6, 1526 CrossRef CAS PubMed.
- G. Repetto, A. del Peso and J. L. Zurita, Nat. Protoc., 2008, 3, 1125 CAS.
- C. M. Sayes, J. D. Fortner, W. Guo, D. Lyon, A. M. Boyd, K. D. Ausman, Y. J. Tao, B. Sitharaman, L. J. Wilson, J. B. Hughes, J. L. West and V. L. Colvin, Nano Lett., 2004, 4, 1881 CrossRef CAS.
- L. R. Hirsch, R. J. Stafford, J. A. Bankson, S. R. Sershen, B. Rivera, R. E. Price, J. D. Hazle, N. J. Halas and J. L. West, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549 CrossRef CAS PubMed.
- E. S. Kaneshiro, M. A. Wyder, Y. P. Wu and M. T. Cushion, Reliability of calcein acetoxy methyl ester and ethidium homodimer or propidium iodide for viability assessment of microbes, J. Microbiol. Methods, 1993, 17, 1–16 CrossRef CAS.
- Y. Luo, W. Chaoming, Y. Qiao, M. Hossain, L. Ma and M. Su, J. Mater. Sci.: Mater. Med., 2012, 23, 2563 CrossRef CAS PubMed.
- L. Braydich-Stolle, S. Hussain, J. J. Schlager and M. C. Hofmann, Toxicol. Sci., 2005, 88, 412 CrossRef CAS PubMed.
- V. A. Fadok, D. L. Bratton, S. C. Frasch, M. L. Warner and P. M. Henson, The role of phosphatidylserine in recognition of apoptotic cells by phagocytes, Cell Death Differ., 1998, 5, 551–562 CAS.
- X. Lu, J. Qian, H. Zhou, Q. Gan, W. Tang, J. Lu, Y. Yuan and C. Liu, Int. J. Nanomed., 2011, 6, 1889 CAS.
- L. H. Reddy, J. S. Adhikari, B. S. R. Dwarakanath, R. K. Sharma and R. R. Murthy, AAPS J., 2006, 8(2), 29 Search PubMed.
- C. D. Bortner, N. B. Oldenburg and J. A. Cidlowski, Trends Cell Biol., 1995, 5, 21 CrossRef CAS.
- B. B. Wolf, M. Schuler, F. Echeverri and D. R. Green, J. Biol. Chem., 1999, 274, 30651 CrossRef CAS PubMed.
- S. Arora, J. Jain, J. M. Rajwade and K. M. Paknikar, Toxicol. Lett., 2008, 179, 93 CrossRef CAS PubMed.
- Y. A. Loannou and F. W. Chen, Nucleic Acids Res., 1996, 24, 992 CrossRef PubMed.
- S. Chandna, Cytometry, Part A, 2004, 61, 127 CrossRef PubMed.
- Z. Darzynkiewicz, D. Galkowski and H. Zhao, Methods, 2008, 44, 250 CrossRef CAS PubMed.
- T. Sun, Y. Yan, Y. Zhao, F. Guo and C. Jiang, PLoS ONE, 2012, 7, e43442 CAS.
- W. Kai, X. Xiaojun, P. Ximing, H. Zhenqing and Z. Qiqing, Nanoscale Res. Lett., 2011, 6, 480 CrossRef PubMed.
- S. Kamada, H. Kusano, H. Fujita, M. Ohtsu, R. C. Koya, N. Kuzumaki and Y. Tsujimoto, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8532 CrossRef CAS.
- A. G. Porter and R. U. Janicke, Cell Death Differ., 1999, 6, 99 CAS.
- L. Foucaud, M. R. Wilson, D. M. Brown and V. Stone, Toxicol. Lett., 2007, 174, 1 CrossRef CAS PubMed.
- C. C. Huang, R. S. Aronstam, D. R. Chen and Y. W. Huang, Toxicol. in Vitro, 2010, 24, 45 CrossRef CAS PubMed.
- M. J. Akhtar, S. Kumar, R. C. Murthy, M. Ashquin, M. I. Khan, G. Patil and I. Ahmad, Toxicol. in Vitro, 2010, 24, 1139 CrossRef CAS PubMed.
- B. C. Heng, X. Zhao, E. C. Tan, N. Khamis, A. Assodani, S. Xiong, C. Ruedl, K. W. Ng and J. S. Loo, Arch. Toxicol., 2011, 85, 1517 CrossRef CAS PubMed.
- R. P. Schins, R. Duffin, D. Höhr, A. M. Knaapen, T. Shi, C. Weishaupt, V. Stone, K. Donaldson and P. J. Borm, Chem. Res. Toxicol., 2002, 15, 1166 CrossRef CAS PubMed.
- I. Papageorgiou, C. Brown, R. Schins, S. Singh, R. Newson, S. Davis, J. Fisher, E. Ingham and C. P. Case, Biomaterials, 2007, 28, 2946 CrossRef CAS PubMed.
- S. Singh, T. Shi, R. Duffin, C. Albrecht, D. van Berlo, D. Höhr, B. Fubini, G. Martra, I. Fenoglio, P. J. Borm and R. P. Schins, Toxicol. Appl. Pharmacol., 2007, 222, 141 CrossRef CAS PubMed.
- K. Bhattacharya, M. Davoren, J. Boertz, R. P. F. Schins, E. Hoffman and E. Dopp, Part. Fibre Toxicol., 2009, 6, 17 CrossRef PubMed.
- P. S. Gilmour, P. H. Beswick, D. M. Brown and K. Donaldson, Carcinogenesis, 1995, 16, 2973 CrossRef CAS PubMed.
- P. S. Gilmour, D. M. Brown, P. H. Beswick, W. MacNee, I. Rahman and K. Donaldson, Environ. Health Perspect., 1997, 105(suppl. 5), 1313 CrossRef CAS.
- V. Stone, H. Johnston and M. J. Clift, IEEE Transactions on NanoBioscience, 2007, 6, 331 CrossRef.
- V. Stone, H. Johnston and R. P. Schins, Crit. Rev. Toxicol., 2009, 39, 613 CrossRef CAS PubMed.
- S. Aula, S. Lakkireddy, A. V. N. Swamy, A. Kapley, K. Jamil, N. R. Tata and K. Hembram, Mater. Res. Express, 2014, 1, 035041 CrossRef.
- S. H. Lee, J. E. Pie, Y. R. Kim, H. R. Lee, S. W. Son and M. K. Kim, Mol. Cell. Toxicol., 2012, 8, 113 CrossRef CAS PubMed.
- S. Alarifi, D. Ali, S. Alkahtani, A. Verma, M. Ahamed, M. Ahmed and H. A. Alhadlaq, Int. J. Nanomed., 2013, 8, 983 Search PubMed.
- Y. Sun, L. W. Oberley and Y. Li, Clin. Chem., 1988, 34, 497 CAS.
- P. Chelikani, I. Fita and P. C. Loewen, Cell. Mol. Life Sci., 2004, 61, 192 CrossRef CAS PubMed.
- B. Fahmy and S. A. Cormier, Toxicol. In Vitro, 2009, 23, 1365 CrossRef CAS PubMed.
- J. Lakritz, C. G. Plopper and A. R. Buckpitt, Anal. Biochem., 1997, 247, 63 CrossRef CAS PubMed.
- C. M. Sayes, A. M. Gobin, K. D. Ausman, J. Mendez, J. L. West and V. L. Colvin, Biomaterials, 2005, 26, 7587 CrossRef CAS PubMed.
- J. R. Arthur, Cell. Mol. Life Sci., 2000, 57, 1825 CrossRef CAS.
- R. Meena, R. Pal, S. N. Pradhan, M. Rani and R. Paulraj, Adv. Mater. Lett., 2012, 3, 459 CAS.
- M. Radu, M. C. Munteanu, S. Petrache, A. I. Serban, D. Dinu, A. Hermenean, C. Sima and A. Dinischiotu, Acta Biochim. Pol., 2010, 57, 355 CAS.
- B. Wu and D. Dong, Trends Pharmacol. Sci., 2012, 33, 656 CrossRef CAS PubMed.
- W. H. Habig, M. J. Pabst and W. B. Jakoby, J. Biol. Chem., 1974, 249, 7130 CAS.
- D. R. Janero, Free Radical Biol. Med., 1990, 9, 515 CrossRef CAS.
- P. V. Asharani, M. P. Hande and S. Valiyaveettil, BMC Cell Biol., 2009, 10, 65 CrossRef CAS PubMed.
- H. Nabeshi, T. Yoshikawa, K. Matsuyama, Y. Nakazato, S. Tochigi, S. Kondoh, T. Hirai, T. Akase, K. Nagano, Y. Abe, Y. Yoshioka, H. Kamada, N. Itoh, S. Tsunoda and Y. Tsutsumi, Part. Fibre Toxicol., 2011, 8, 1 CrossRef CAS PubMed.
- B. N. Ames, W. E. Durston, E. Yamasaki and F. D. Lee, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 2281 CrossRef CAS.
- H. R. Kim, Y. J. Park, D. Y. Shin, S. M. Oh and K. H. Chung, Environ. Health Toxicol., 2013, 28, e2013003 Search PubMed.
- R. Landsiedel, M. D. Kapp, M. Schulz, K. Wiench and F. Oesch, Mutat. Res., 2009, 681, 241 CAS.
- K. Chu and M. J. Tsai, Diabetes, 2005, 54, 1064 CrossRef CAS.
- X. Kong, G. R. Hellermann, W. Zhang, P. Jena, M. Kumar, A. Behera, S. Behera, R. Lockey and S. S. Mohapatra, Allergy, Asthma, Clin. Immunol., 2008, 4, 95 CrossRef CAS PubMed.
- M. J. Holden and L. Wang, Quantitative Real Time PCR: Fluorescent Probe Options and Issues, in Standardization and Quality Assurance in Fluorescence Measurements: Bioanalytical and Biomedical Applications, in Fluorescence: Methods and Applications, ed. U. Resch-Genger, Springer-Verlag, Berlin, 2008, vol. 6, pp. 489–508 Search PubMed.
- M. J. Akhtar, M. Ahamed, S. Kumar, M. A. M. Khan, J. Ahmad and S. A. Alrokayan, Int. J. Nanomed., 2012, 7, 845 CAS.
- Y. Zhang, Z. Hu, M. Ye, Y. Pan, J. Chen, Y. Luo, Y. Zhang, L. He and J. Wang, Eur. J. Pharm. Biopharm., 2007, 66, 268 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, J. D. Clogston, B. W. Neun, J. B. Hall, A. K. Patri and S. E. McNeil, Nano Lett., 2008, 8, 2180 CrossRef CAS PubMed.
- S. M. Moghimi, J. Controlled Release, 2014, 190, 556 CrossRef CAS PubMed.
- M. Vittaz, D. Bazile, G. Spenlehauer, T. Verrecchia, M. Veillard, F. Puisieux and D. Labarre, Biomaterials, 1996, 17, 1575 CrossRef CAS.
- J. Szebeni, L. Baranyi, S. Savay, J. Milosevits, M. Bodo, R. Bunger and C. R. Alving, Methods Enzymol., 2003, 373, 136 CAS.
- S. M. Chowdhury, S. Kanakia, J. D. Toussaint, M. D. Frame, A. M. Dewar, K. R. Shroyer, W. Moore and B. Sitharaman, Sci. Rep., 2013, 3, 2584 Search PubMed.
- I. Hamad, A. C. Hunter and S. M. Moghimi, J. Controlled Release, 2013, 170, 167 CrossRef CAS PubMed.
- A. N. Ilinskaya and M. A. Dobrovolskaia, Nanomedicine, 2013, 8, 969 CrossRef CAS PubMed.
- H. Sahli, J. Tapon-Bretaudière, A. M. Fischer, C. Sternberg, G. Spenlehauer, T. Verrecchia and D. Labarre, Biomaterials, 1997, 18, 281 CrossRef CAS.
- B. W. Neun and M. A. Dobrovolskaia, Methods Mol. Biol., 2011, 697, 225 CAS.
- T. T. W. Shwe, S. Yamamoto, M. Kakeyama, T. Kobayashi and H. Fujimaki, Toxicol. Appl. Pharmacol., 2005, 209, 51 CrossRef PubMed.
- C. M. Sayes, K. L. Reed and D. B. Warheit, Toxicol. Sci., 2007, 97, 163 CrossRef CAS PubMed.
- P. Mantecca, G. Sancini, E. Moschini, F. Farina, M. Gualtieri, A. Rohr, G. Miserocchi, P. Palestini and M. Camatini, Toxicol. Lett., 2009, 189, 206 CrossRef CAS PubMed.
- J. Leszczynski, Nat. Nanotechnol., 2010, 5, 633 CrossRef CAS PubMed.
- P. P. Adiseshaiah, J. B. Hall and S. E. McNeil, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2, 99 CrossRef CAS PubMed.
- A. Monopoli, A. S. Pitek, I. Lynch and K. A. Dawson, Formation and Characterization of the Nanoparticle–Protein Corona, in Nanomaterial Interfaces in Biology : Methods and Protocols, ed. P. Bergese and K. Hamad-Schifferli, Springer Humana press, New York, 2013, vol. 1025, pp. 137–155 Search PubMed.
- H. R. Kim, K. Andrieux, C. Delomenie, H. Chacun, M. Appel, D. Desmaële, F. Taran, D. Georgin, P. Couvreur and M. Taverna, Electrophoresis, 2007, 28, 2252 CrossRef CAS PubMed.
- D. Lorusso, A. Di Stefano, V. Carone, A. Fagotti, S. Pisconti and G. Scambia, Ann. Oncol., 2007, 18, 1159 CrossRef CAS PubMed.
- N. Yokomichi, T. Nagasawa, A. Coler-Reilly, H. Suzuki, Y. Kubota, R. Yoshioka, A. Tozawa, N. Suzuki and Y. Yamaguchi, Hum. Cell, 2013, 26, 8 CrossRef CAS PubMed.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703 CrossRef CAS PubMed.
- T. Daemen, G. Hofstede, M. T. Ten Kate, I. A. J. M. Bakker-Woudenberg and G. L. Scherphof, Int. J. Cancer, 1995, 61, 761 CrossRef PubMed.
- J. P. Plard and D. Bazile, Colloids Surf., B, 1999, 16, 173 CrossRef CAS.
- I. Hamad, A. Christy Hunter, K. J. Rutt, Z. Liu, H. Dai and S. Moein Moghimi, Mol. Immunol., 2008, 45, 3797 CrossRef CAS PubMed.
- T. R. Downs, M. E. Crosby, T. Hu, S. Kumar, A. Sullivan, K. Sarlo, B. Reeder, M. Lynch, M. Wagner, T. Mills and S. Pfuhler, Mutat. Res., 2012, 745, 38 CAS.
- S. Ivanov, S. Zhuravsky, G. Yukina, V. Tomson, D. Korolev and M. Galagudza, Materials, 2012, 5, 1873 CrossRef CAS PubMed.
- M. A. K. Abdelhalim and B. M. Jarrar, J. Nanobiotechnol., 2012, 10, 5 CrossRef CAS PubMed.
- A. M. Prodan, S. L. Iconaru, C. S. Ciobanu, M. C. Chifiriuc, M. Stoicea and D. Predoi, J. Nanomater., 2013, 2013, 587021 Search PubMed.
- K. M. A. Hassanin, S. H. A. El-Kawi and K. S. Hashem, Int. J. Nanomed., 2013, 8, 1713 Search PubMed.
- J. S. Kim, J. H. Sung, J. H. Ji, K. S. Song, J. H. Lee, C. S. Kang and I. J. Yu, Saf. Health Work, 2011, 2, 34 CAS.
- Z. Chen, Y. Wang, T. Ba, Y. Li, J. Pu, T. Chen, Y. Song, Y. Gu, Q. Qian, J. Yang and G. Jia, Toxicol. Lett., 2014, 226, 314 CrossRef CAS PubMed.
- L. H. Reddy, P. E. Marque, C. Dubernet, S. L. Mouelhi, D. Desmaele and P. Couvreur, Drug Metab. Dispos., 2008, 325, 484 CAS.
- M. Li, K. T. Al-Jamal, K. Kostarelos and J. Reineke, ACS Nano, 2010, 4, 6303 CrossRef CAS PubMed.
- P. Espié, D. Tytgat, M. L. Sargentini-Maier, I. Poggesi and J. B. Watelet, Drug Metab. Rev., 2009, 41, 391 CrossRef PubMed.
- L. MacCalman, C. L. Tran and E. Kuempel, J. Phys.: Conf. Ser., 2009, 151, 012028 CrossRef.
- C. L. Tran, E. D. Kuempel and V. Castranova, Ann. Occup. Hyg., 2002, 46(suppl 1), 14 CrossRef.
- D. P. Lankveld, A. G. Oomen, P. Krystek, A. Neigh, A. Troost-de Jong, C. W. Noorlander, J. C. Van Eijkeren, R. E. Geertsma and W. H. De Jong, Biomaterials, 2010, 31, 8350 CrossRef CAS PubMed.
- H. A. Lee, T. L. Leavens, S. E. Mason, N. A. Monteiro-Riviere and J. E. Riviere, Nano Lett., 2009, 9, 794 CrossRef CAS PubMed.
- M. Li, Z. Panagi, K. Avgoustakis and J. Reineke, Int. J. Nanomed., 2012, 7, 1345 CAS.
- P. Lin, J. W. Chen, L. W. Chang, J. P. Wu, L. Redding, H. Chang, T. K. Yeh, C. S. Yang, M. H. Tsai, H. J. Wang, Y. C. Kuo and R. S. Yang, Environ. Sci. Technol., 2008, 42, 6264 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |