
PtFe/nitrogen-doped graphene for high-performance electrooxidation of formic acid with composition sensitive electrocatalytic activity
Yanfang Sunab,
Ting Zhoua,
Qingyan Pana,
Xiao Zhang*a and
Jinxue Guo*a
aKey Laboratory of Sensor Analysis of Tumor Marker (Ministry of Education), Key Laboratory of Rubber-plastics (Ministry of Education), Laboratory of Inorganic Synthesis and Applied Chemistry, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, PR China. E-mail: zhx1213@126.com; gjx1213@126.com; Tel: +86 532 84022681
bCollege of Science and Technology, Agricultural University of Hebei, Cangzhou 061100, China
First published on 24th June 2015
Abstract
With the aim to pursue novel high-performance electrocatalysts for fuel cells, a simple synthesis strategy, which consists of hydrothermal reaction followed by solid state reduction by H2, is developed to prepare a series of PtxFe100−x/N-doped graphene nanocomposites with controllable Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe compositions. The morphology, microstructure, and composition of the samples are systematically characterized with transmission electron microscopy, scanning electron microscopy, energy dispersive spectroscopy, powder X-ray diffraction, X-ray photoelectron spectroscopy and Raman spectroscopy. The effects of nitrogen doping and alloying with Fe, as well as their synergistic interactions, on the improvement of the catalytic performance are well revealed by using the present samples as catalysts for formic acid electrooxidation. Additionally, the composition sensitive catalytic activity and stability of these catalysts for formic acid electrooxidation are probed and the optimum Pt:Fe ratio is presented. The optimum sample possesses both enhanced electrochemical performance and a reduced dosage of the noble metal, making it a promising candidate for fuel cell applications.
:Fe compositions. The morphology, microstructure, and composition of the samples are systematically characterized with transmission electron microscopy, scanning electron microscopy, energy dispersive spectroscopy, powder X-ray diffraction, X-ray photoelectron spectroscopy and Raman spectroscopy. The effects of nitrogen doping and alloying with Fe, as well as their synergistic interactions, on the improvement of the catalytic performance are well revealed by using the present samples as catalysts for formic acid electrooxidation. Additionally, the composition sensitive catalytic activity and stability of these catalysts for formic acid electrooxidation are probed and the optimum Pt:Fe ratio is presented. The optimum sample possesses both enhanced electrochemical performance and a reduced dosage of the noble metal, making it a promising candidate for fuel cell applications.
Introduction
Increasing demands for renewable and clean energy sources with high energy density and a low operating temperature have aroused intensive research interest in the development of high-efficiency fuel cells. To satisfy the strict requirement of commercial fuel cells with a high current density, electrocatalysts which can highly efficiently oxidize small organic molecules are desired to be achieved. Among the highly efficient catalysts of the noble metal family, Pt is the preferred catalyst for fuel cells due to its specific catalytic ability to facilitate both the oxidation and reduction reactions.1 At present, a state-of-art fuel cell requires 0.5 mg Pt per cm2 of electrode area.2 So the production and application of fuel cells require a relatively high consumption of Pt, which is scarce and expensive.3 In order to decrease the usage amount of Pt and promote the commercialization of Pt-based fuel cells, Pt-based catalysts with enhanced catalytic activities are desired to be achieved. To meet this requirement, two major strategies can be adopted: exploring high-efficiency catalysts with a lower Pt content, and developing novel alternative support materials with better support performance to replace the traditional carbon black supports due to their bulk structure and low surface area.4As for the first solution, the alloying of Pt with another metal (M) is believed to be an effective way to reduce the consumption of Pt, where M is usually one of the transition metals such as Fe, Co, Ni, Cu, Ti, Pb, Zn, Cd, Hg, Sn, Ru, and Au.5–22 As demonstrated in the literature, the addition of transition metals into Pt-based alloy catalysts can not only promote their electrocatalytic activity, but also improve the tolerance to CO poisoning.23,24 On the other hand, graphene has been widely used as an alternative and effective support material to boost the catalytic performance of Pt, which exhibits a unique structure and interesting physical properties such as huge surface area, a flexible 2D form, good electrical and thermal conductivities, charge transport mobility, and good chemical stability.13–15,25 In our previous report, a FePt nanoalloy anchored graphene composite was presented and demonstrated excellent electrocatalytic activity in formic acid and methanol oxidation.9 Recently, numerous reports have predicated that, doping graphene with heteroatoms, particularly with N atoms, could effectively tailor its intrinsic electronic characteristics.26–29 For instance, N-doped graphene (NG) shows a different spin density and charge distribution compared with pristine graphene, due to the influence of the nitrogen dopants on their neighboring carbon atoms.28–31 It will introduce an “activation region” on the graphene surface, which can participate in catalytic reactions directly and anchor metal nanoparticles used in the catalytic reaction. This kind of material has currently been studied extensively for applications in energy conversion and storage, such as in supercapacitors, lithium-ion batteries, and especially oxygen reduction reactions.32–36 So it is worthwhile exploring the influence of NG on the catalytic oxidation activity of Pt-based catalysts for fuel cells.
For fuel cells, the liquid phases of formic acid and methanol are two more attractive energy sources than gaseous or liquid hydrogen due to the ease of handling, transportation and storage.37 In particular, a great deal of research efforts have been focused on direct formic acid fuel cells (DFAFC), because the electrooxidation of formic acid occurs at lower positive potentials than methanol, and the crossover of formic acid through the polymer membrane is lower than for methanol.38,39 Recently, some NG supported Pt-based catalysts have been synthesized and their electrooxidation performance for methanol has been measured,20–22,40,41 but studies on formic acid oxidation are rare. Therefore, it would be of great interest to evaluate the electrocatalytic performance of NG supported Pt-based catalysts in formic acid oxidations.
In this manuscript, Pt–Fe alloy nanoparticles anchored on NG sheets are prepared as electrocatalysts for the catalytic oxidation of formic acid. Considering that the alloy composition is also an extremely important factor with respect to the electrocatalytic properties,11,42,43 PtxFe100−x/NG catalysts with diverse composition ratios are presented and the effect of composition on the catalytic activity is revealed systematically. The results show that, the as-fabricated PtxFe100−x/NG catalysts exhibit enhanced electrocatalytic activity in formic acid oxidations and better catalytic stability, due to the contributions from the alloying effect of PtFe and the synergistic interactions with the NG sheets. The Pt43Fe57/NG catalyst delivers the highest catalytic activity for formic acid oxidation.
Experimental
Synthesis of the graphene oxides
All reagents were obtained from Sinopharm Chemical Reagent Co., Ltd. They were of analytical grade, and were used as purchased without further purification. Graphene oxides (GOs) are prepared by oxidizing natural graphite according to a modified Hummers method.44 Briefly, graphite powder (2 g) and NaNO3 (1 g) are added into 50 mL of concentrated H2SO4 (98%) for 30 min of vigorous stirring at 5 °C. KMnO4 (0.3 g) is added into the mixture for another 30 min of stirring at 10 °C. KMnO4 (7 g) is added into the mixture gradually within 1 h, in order to keep the temperature below 20 °C. Subsequently, the mixture is stirred at 35 ± 5 °C for 2 h to obtain a brown dispersion solution. After 90 mL of distilled water is poured into the dispersion, the temperature is increased quickly and kept above 90 °C for 15 min. Finally, 55 mL of distilled water and 10 mL of 30% H2O2 solution are poured into the mixture to terminate the reaction. The resultant light yellow dispersion is filtered, and then is washed with 1 M HCl aqueous solution and a huge amount of distilled water in sequence. The obtained black powders are exfoliated with ultrasonication in water for more than 1 h to prepare homogeneous brown-black GO dispersions with a concentration of 10 mg mL−1.Synthesis of the catalysts
The synthesis of the Pt79Fe21/N-doped graphene composite (Pt79Fe21/NG) can be described as follows: firstly, 12 mg of K4Fe(CN)6·3H2O and 150 mg of urea are dissolved in 24 mL of deionized water. After mixing with 6 mL of GO solution (5 mg mL−1), the resultant solution is transferred into a 40 mL autoclave and maintained at 180 °C for 4 h. The products are collected using centrifugation and then washed with absolute ethanol and water in turn before vacuum drying at 60 °C. Subsequently, 60 mg of the hydrothermal product is dispersed in 60 mL of H2PtCl6·3H2O solution (0.5 mg mL−1). The dispersion is kept under ultrasonic irradiation for 30 min and then evaporated at 80 °C under vigorous stirring. Finally, the dried black powder is loaded into a tube furnace and heat-treated at 120 °C for 2 h in a H2/Ar (volume ratio of 5/95) atmosphere with a heating rate of 1 °C min−1 to obtain the final product of Pt79Fe21/NG.The synthesis procedures of the other PtxFe100−x/NG catalysts are the same as for the preparation of Pt79Fe21/NG except for the different doses of K4Fe(CN)6·3H2O as listed in Table 1. For comparison, Pt/NG, PtFe/graphene (PtFe/G), and Pt/G catalysts were prepared via the same synthetic procedure mentioned above by varying the reactants as listed in Table 1.
| Samples | K4Fe(CN)6·3H2O | H2PtCl6·3H2O | Urea |
|---|---|---|---|
| Pt79Fe21/NG | 12 | 30 | 150 |
| Pt60Fe40/NG | 39 | 30 | 150 |
| Pt43Fe57/NG | 60 | 30 | 150 |
| Pt27Fe73/NG | 120 | 30 | 150 |
| Pt20Fe80/NG | 180 | 30 | 150 |
| Pt16Fe84/NG | 240 | 30 | 150 |
| Pt/NG | 0 | 30 | 150 |
| Pt79Fe21/G | 12 | 30 | 0 |
| Pt/G | 0 | 30 | 0 |
Characterization of the catalysts
The crystal structures of the as-prepared samples were characterized using powder X-ray powder diffraction (XRD, Philips X’-pert X-ray diffractometer). Scanning electron microscopy (SEM) and energy dispersive spectroscopy (EDS) were performed with a JEOL JSM-7500F. Transmission electron microscopy (TEM) was performed on a JEOL, JEM-2100. Fourier transform infrared (FTIR) spectra were obtained using a Nicolet Magna-IR750. Raman spectroscopy was performed using a confocal microprobe Raman system (LabRam-e010, 632 nm as the excitation source). X-ray photoelectron spectroscopy (XPS) measurements were carried out on a RBD upgraded PHI-5000c ESCA system (Perkin Elmer) with Mg Kα radiation (hν = 1253.6 eV).Electrochemical measurements
All the electrochemical tests were performed on a CHI600D (CH Instruments, Shanghai, China) using a conventional three-electrode cell at room temperature. A modified glassy carbon electrode (GCE), a saturated calomel electrode (SCE), and a plate wire were used as the working, reference, and counter electrode, respectively. A mixture of 2 mg of catalyst, 0.5 mL of Nafion solution (0.05 wt%), and 0.5 mL of ethanol was ultrasonically dispersed to prepare a homogeneous catalyst ink. One portion of the catalyst ink (3 μL) was casted onto the pretreated GCE surface to fabricate the modified GCE electrode, which was used as the working electrode for the electrochemical measurements. The catalytic performance of the catalysts for electrooxidation of formic acid was measured with cyclic voltammetry (CV), which was performed between −0.2 V and 1.2 V at a scan rate of 50 mV s−1 in a 0.5 M H2SO4 solution containing 1 M HCOOH. Chronoamperometric curves were obtained at a fixed potential of 0.3 V for 10 min. Prior to the electrochemical tests, the electrolyte solutions were deaerated by bubbling with N2 for 15 min.Results and discussion
Characterization of the samples
The as-obtained samples are characterized using TEM techniques. Fig. 1a shows the TEM image of the Pt/G composite. Pt particles are aggregated and located unevenly on the surface of the graphene sheets with a broad particle size distribution. As shown in Fig. 1b, the PtFe alloy particles show a larger particle size and deliver worse dispersion states than Pt. In contrast, the alloy nanoparticles in the case of Pt79Fe21/NG are dispersed uniformly and densely on the NG sheet surface (Fig. 1c). As observed from the magnified TEM image (Fig. 1d), the Pt79Fe21 nanoalloys are monodisperse with a narrow size distribution. The size distribution of the PtFe alloy nanoparticles on the NG sheets is evaluated statistically by measuring the diameters of two hundred particles from the TEM images and the mean size is around 3.7 nm (inset in Fig. 1c). It has been reported that, the nitrogen dopants in NG sheets can lead to localized defects and promote the graphene chemical activation for enhanced interactions with nanoparticles, thus facilitating their assembly and benefiting their dispersion.20–22,33,40 Hence, it can be concluded that the improved dispersity of the Pt79Fe21 nanoalloys should be ascribed to the presence of the N-doped sites in the NG sheets, which act as bonding sites to in situ anchor the nanoparticle precursors.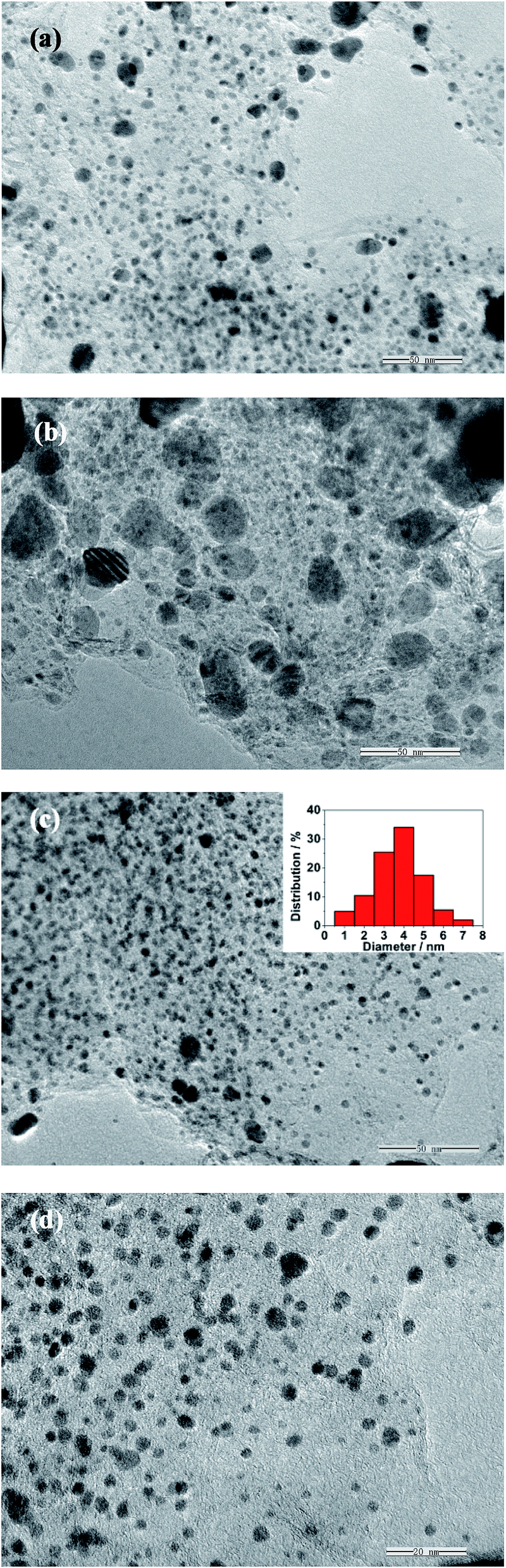 | ||
| Fig. 1 TEM images of (a) Pt/G, (b) PtFe/G, (c) and (d) Pt79Fe21/NG. | ||
The morphology of the as-obtained Pt79Fe21/NG catalyst is investigated with SEM and is shown in Fig. 2a. The representative wrinkled layer structure of the graphene sheets can be observed clearly. EDS is an effective technique to characterize the composition of a composite. As shown in the EDS spectrum of Pt79Fe21/NG (Fig. 2b), Pt, Fe, C, N, and O elements are observed. The Fe and Pt elements can be assigned to the PtFe alloys. The C element corresponds to graphene. The observation of the N peak suggests the existence of N elements in the composite catalyst. The small amount of O can be ascribed to the residual oxygen-containing functional groups of RGO.9 The inset table of Fig. 2b shows that the weight ratio of C:Pt:Fe:N was determined to be 53.82:26.31:2.08:3.97 with a corresponding atomic ratio of 77.25:2.32:0.64:4.89. The EDS mapping of C, Pt, Fe, and N are presented in Fig. 2c–f, respectively. The elements of C and Pt are homogeneously and densely distributed. However, the Fe and N elements are distributed evenly and sparsely, which is attributed to their lower content. Fig. 2g shows the EDS elemental mapping images of Pt (blue points) and Fe (green points). It can be clearly seen that, the Pt and Fe elements accompany each other, indicating the formation of a PtFe alloy. The NG sheets are almost fully enveloped by the uniformly and densely dispersed PtFe nanoalloys to form a sandwich-like composite structure, which is consistent with the TEM measurements.
 | ||
| Fig. 2 (a) SEM image of Pt79Fe21/NG. (b) EDS spectrum of Pt79Fe21/NG. Inset shows the atomic and weight ratio of Fe, Pt, C, and N elements. The EDS mapping of C (c), Pt (d), Fe (e), and N (f). (g) EDS elemental mapping image of the Pt (blue points), Fe (green points), and C (red points) in Pt79Fe21/NG. | ||
To further characterize the elemental composition and nitrogen bonding configurations in Pt79Fe21/NG, XPS measurements were performed. As shown in Fig. 3a, the survey spectrum of Pt79Fe21/NG indicated the existence of C, O, N, Fe, and Pt elements, which is consistent with the EDS measurements. A high-resolution N 1s XPS spectrum is shown in Fig. 3b, which is powerful for probing the nature of the N functionalities. Five deconvoluted peaks can be observed from it, indicating the presence of pyridinic N and Fe–Nx (298.3 eV), amino N (399.2 eV), pyrrolic N (400.2 eV), graphitic N (401.2 eV), and oxidized N (403.0 eV).49,50 The existence of Fe–Nx reveals the connection between the doped N and the metal Fe, which explains the overlapped elemental mapping of Fe and N, and benefits the uniform distribution of alloy nanoparticles on the surface of the NG nanosheets. Among these N functionalities, graphitic N, which represents the N atoms doped into the graphitic basal plane, is generally believed to be responsible for the enhanced catalytic activity of N-doped carbon materials.26–29,49 Therefore, the presence of graphitic N in the Pt79Fe21/NG catalyst will undoubtedly boost its catalytic performance.
 | ||
| Fig. 3 XPS spectra of Pt79Fe21/NG: (a) survey spectrum and (b) high-resolution N 1s spectra. | ||
XRD patterns are obtained to determine the composition and crystal structure of the as-prepared GO, NG, Pt/NG and Pt79Fe21/NG. As shown in Fig. 4a, the representative diffraction peak of (001) for the GO sheets is observed at a 2θ of 9.3°, corresponding to a d-spacing of 0.95 nm. A broad (002) diffraction peak appears at a 2θ of ∼25.2° in the XRD pattern of NG, indicating that the GO nanosheets have been successfully reduced to NG sheets after the hydrothermal reaction with urea.15,45 Besides the NG peak, there are three other peaks at 40°, 46°, and 68° in both the Pt/NG and Pt79Fe21/NG nanocomposites, which match well with the (111), (200), and (220) planes of a face-centered cubic structure of Pt or PtFe nanoalloys (JCPDS card no. 04-0802). In the XRD pattern of Pt79Fe21/NG, no other impurity peaks corresponding to metallic Fe or its oxides are detected, confirming its high phase purity. Interestingly, in the case of Pt79Fe21/NG, the characteristic peaks shift slightly to higher 2θ values with respect to the corresponding peaks for the Pt/NG. Fig. 4b shows magnified (111) peaks from the XRD patterns of Pt/NG and Pt79Fe21/NG. The position of the (111) peak of Pt79Fe21/NG is located at a higher 2θ value than that of Pt/NG, which can be attributed to the contraction of the lattice constants, indicating the formation of a Pt–Fe alloy.9,15
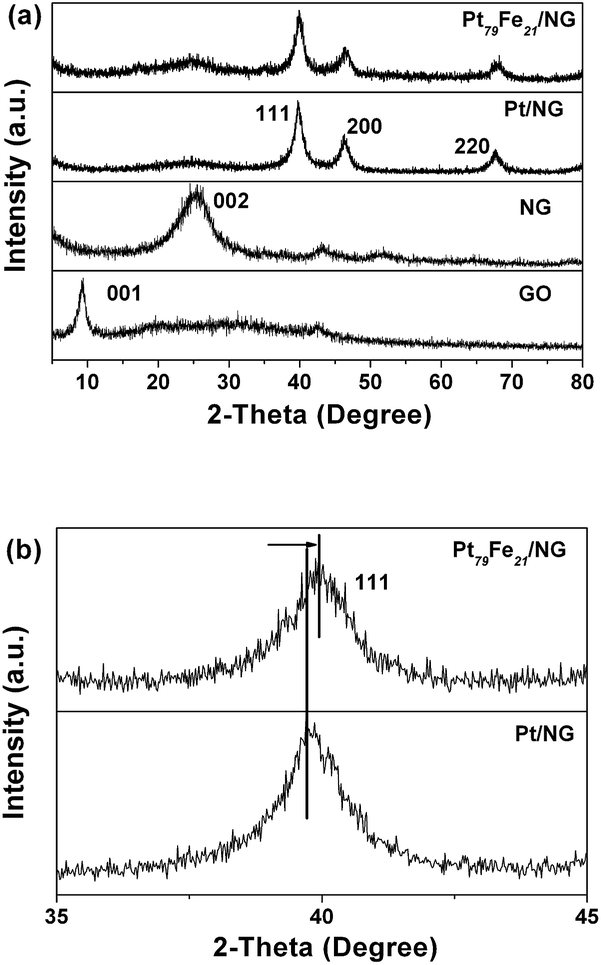 | ||
| Fig. 4 (a) XRD patterns of GO, NG, Pt/NG and Pt79Fe21/NG; (b) magnified XRD (111) peaks of Pt/NG and Pt79Fe21/NG. | ||
Raman spectroscopy is a very useful tool for characterizing structural defects and the doping level of graphene.20,22,28 Fig. 5 shows the Raman spectra of the GO, NG, and Pt79Fe21/NG. Two representative bands of D bands (corresponding to structural defects and disorder in graphene) and G bands (corresponding to the optical mode vibration of two neighboring carbon atoms) are the predominant features in the spectra of all three samples. Specifically, the intensity ratio of the D and G bands (ID/IG) is directly proportional to the defects and can be used to characterize the nitrogen doping in graphene.28 Compared with GO, the NG sheets show a red-shift of the D band and an increased ID/IG value from 0.94 to 1.12, which should be assigned to the doping of nitrogen into the graphene, as well as the structural and edge defects in graphene. After anchoring with PtFe nanoalloys, the Pt79Fe21/NG also displays a slightly red-shifted D band compared with GO due to the nitrogen doping.20,22,28 It delivers a further increased ID/IG of 1.27, suggesting interaction between the PtFe nanoalloys and NG substrates.46
 | ||
| Fig. 5 Raman spectra of the GO, NG, and Pt79Fe21/NG. | ||
Electrochemical measurements
Cyclic voltammograms recorded for the Pt/G, Pt79Fe21/G, and Pt79Fe21/NG in N2-saturated 0.5 M H2SO4 at a scan rate of 50 mV s−1 are shown in Fig. 6. Pristine graphene and NG electrodes are also tested for comparison. No obvious peak current can be observed for graphene and NG. The other Pt-based samples deliver similar voltammetric features. A typical pair of broad current peaks related to hydrogen adsorption–desorption are observed between −0.2 and 0.0 V. The anodic peak at about 0.4 V corresponds to the oxidation reaction of Pt (Pt + H2O → Pt–(OH)ad + H+ + e− → Pt–Oad + H+ + e−). The reversible reduction reaction of the Pt oxides can be observed at about 0.3 V in the cathodic scan. Such voltammetric features have been reported for other Pt based alloys, which can be assigned to the formation of a Pt skin on the catalyst surface.9,22,42,47 Among these three kinds of catalysts, Pt79Fe21/NG shows the most prominent reduction peak currents for the Pt oxides and hydrogen adsorption–desorption region, which might result from the enhanced synergistic effects between the Pt–Fe alloy and N-G.22 Interestingly, the Pt79Fe21/G catalyst shows a slightly positive-shifted reduction peak for the Pt oxides in comparison with Pt/G, due to the presence of the Fe in the alloy which decreases the desorption free energy of Pt–OH, Pt–O, or Pt–O2 species. It will lower the adsorption strength of the adsorbed oxygen species on the Pt in the alloy and provide more facile oxidation sites for the intermediates.47 A further positive shift of the reduction peak is observed for the Pt79Fe21/NG, which should be attributed to the N doping. | ||
| Fig. 6 CV curves of the GO, NG, Pt/G, Pt79Fe21/G, and Pt79Fe21/NG catalysts in 0.5 M H2SO4. | ||
It is believed that the doping of nitrogen into graphene could introduce atomic charge density and asymmetry in the spin density of the graphene network, thus facilitating charge transfer between the graphene support and the anchoring metals.22,26–31 So when NG is used as the support for Pt-based catalysts, it can evidently enhance the interactions between the catalytic metals and carbon substrate, thus improving the catalytic activity for formic acid electrooxidation. With the aim to reveal the enhanced catalytic activity of the presented PtxFe100−x/NG catalysts, electrochemical formic acid oxidation reactions using the GO, NG, Pt/G, Pt/NG, Pt79Fe21/G, and Pt79Fe21/NG catalysts were evaluated using CV in 0.5 M H2SO4 containing 1 M HCOOH. It is well known that HCOOH electrooxidation on Pt catalysts usually has a dual-pathway mechanism: (1) the direct pathway to CO2 via a dehydrogenation process that involves a reactive intermediate of adsorbed formate (HCOOad), and (2) the indirect pathway through a dehydration reaction with the formation of a poisoning intermediate COad.17,43
| HCOOH → HCOOad + H+ + e− → CO2 + 2H+ + 2e− | (1) |
| HCOOH → COad + H2O → CO2 + 2H+ + 2e− | (2) |
Notably, as shown in Fig. 7, there is almost no peak current for the GO and NG modified electrodes, suggesting that they show no catalytic activity for formic acid electrooxidation. In contrast with this, all the Pt-based catalysts deliver a distinct peak current and show similar voltammetric features. Only one peak at around 0.7 V, which corresponds to the oxidation of CO generated by the dehydration of HCOOH, is observed for the positive scan of the CV curve.42 The oxidation current peak for direct oxidation of HCOOH into CO2 is indistinct. The peak corresponding to the oxidation of the intermediate species formed during the formic acid oxidation is observed in the negative scan of the CV curves. These voltammetric features indicate that formic acid electrooxidation on these catalysts occurs mainly through the indirect pathway. The oxidation peak current for the negative scan of Pt/G is 25 mA mg−1. In contrast with this, Pt/NG (62 mA mg−1) and Pt79Fe21/G (111 mA mg−1) show higher peak currents due to the nitrogen doping and the alloying with Fe, respectively. The Pt79Fe21/NG catalyst delivers the highest value of 186 mA mg−1 among these four samples, corresponding to the enhanced synergistic effects between the Pt–Fe alloy and NG. It is worth noting that the peaks located in the negative scan are observed at ∼0.27 and 0.29 V for Pt/NG and Pt79Fe21/NG, respectively, while the peaks of Pt/G and Pt79Fe21/G locate at ∼0.37 and 0.43 V, respectively. This obvious negative shift of the oxidation peak should be assigned to the nitrogen doping, which is beneficial for formic acid electrooxidation. Noting that, the oxidation peak of Pt/NG in the negative scan is located at ∼0.27 V, showing an obvious negative shift compared with that of Pt/G, which should be ascribed to the nitrogen doping of graphene. The comparison of the peak positions between Pt79Fe21/NG and Pt79Fe21/G also confirms this conclusion. So it can be concluded that NG substrates can reduce the oxygen potential of the composite catalyst for formic acid indirect electrooxidation, thus improving the catalytic activities of the composite catalyst.
 | ||
| Fig. 7 CV curves of the GO, NG, Pt/G, Pt/NG, Pt79Fe21/G, and Pt79Fe21/NG catalysts in 0.5 M H2SO4 containing 1 M HCOOH. | ||
Fig. 8 allows comparison of the catalytic activity of the PtxFe100−x/NG catalysts to elucidate the composition sensitivity of the formic acid oxidation. Initially, with the increasing Fe composition in the alloy, the oxidation peak current of the negative scan increases from 186 mA mg−1 for Pt79Fe21/NG to 421 mA mg−1 for Pt60Fe40/NG and reaches up to the highest value of 603 mA mg−1 for Pt43Fe57/NG, which is ∼24 times that of Pt/G. After that, the catalytic activity of PtxFe100−x/NG decreases with further increase of the Fe component. The oxidation peak current values are 281 and 97 mA mg−1 for Pt27Fe73/NG and Pt20Fe80/NG, respectively. When the Fe composition increases up to 84%, the as-obtained Pt16Fe84/NG nanocomposite almost shows no detectable catalytic activity for formic acid oxidation.
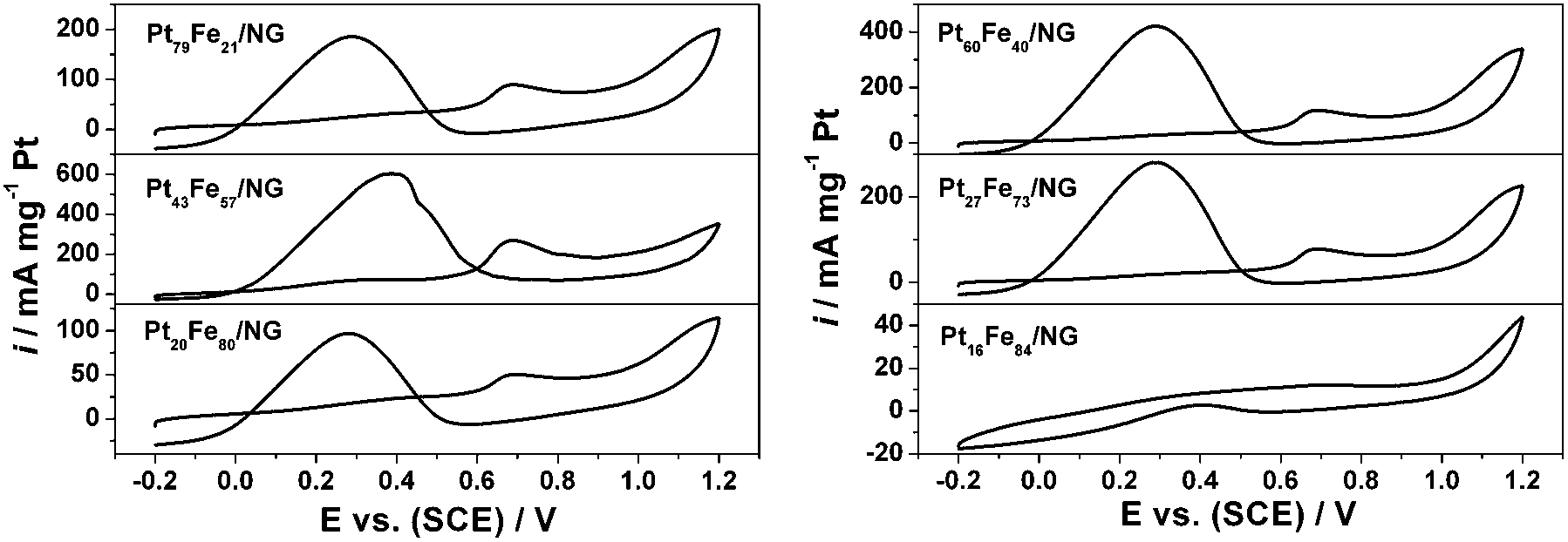 | ||
| Fig. 8 CV curves of the PtxFe100−x/NG catalysts in 0.5 M H2SO4 solution containing 1 M HCOOH at a scan rate of 50 mV s−1. | ||
Fig. 9 presents the TEM images of Pt60Fe40/NG, Pt43Fe57/NG, Pt27Fe73/NG, Pt20Fe80/NG, and Pt16Fe84/NG. Except for Pt16Fe84/NG, the alloy nanoparticles in the other four kinds of catalysts are dispersed on the NG sheets with a narrow size distribution, which is similar to that of Pt79Fe21/NG. As for Pt16Fe84/NG, a few large particles are observed, due to the high ratio of Fe in the alloy. So it can be concluded that, the presented PtxFe100−x/NG catalysts show obvious composition sensitive activities for formic acid electrooxidation. The optimum composition in terms of the Pt:Fe atomic ratio is obtained as about 1:1 in our presented study. It is believed that, under certain optimum compositions, the surface area of the Pt skin is maximized to absorb HCOOH and the second metal supplies enough surface sites to promote the effective oxidative removal of poisonous intermediates for Pt-based bimetallic alloy electrocatalysts. This optimum composition ratio is consistent with previous results for FePt and PtRu catalysts.42,48
 | ||
| Fig. 9 TEM images of (a) Pt60Fe40/NG, (b) Pt43Fe57/NG, (c) Pt27Fe73/NG, (d) Pt20Fe80/NG, and (e) Pt16Fe84/NG. | ||
To further compare the catalytic activity and durability of the PtxFe100−x/NG catalysts, chronoamperometry tests were conducted and the results are shown in Fig. 10. The catalytic performances of the GO and NG were also investigated for comparison. In accordance with the CV results in Fig. 7, the GO and NG modified electrodes show no catalytic activity for formic acid oxidation. As observed from Fig. 10, the catalytic stability of Pt16Fe84/NG is very poor and the activity can only be maintained for several seconds. Pt20Fe80/NG delivers a similar curve to the results reported in most literature, which drops rapidly at the primary stage and then decays slowly to a limiting value. As for the Pt79Fe21/NG, Pt60Fe40/NG, Pt43Fe57/NG and Pt27Fe73/NG catalysts, the chronoamperometric curves show an interesting increase at the first stage, and then show analogous features to Pt20Fe80/NG. This exceptional increase should be attributed to the indirect oxidation pathway of formic acid electrooxidation on these catalysts. In the primary oxidation process, the intermediate species have not formed. So the oxidation current density is low. With the reaction of formic acid oxidation going on, the amount of intermediate species rises, thus inducing the increase of current density at the first stage. This result further confirms the indirect oxidation pathway for formic acid electrooxidation on PtxFe100−x/NG catalysts. One can see from these curves that Pt43Fe57/NG delivers the highest start current density, which is consistent with the CV results presented in Fig. 8. But after ∼200 s, the curve of Pt43Fe57/NG is overlapping with that of Pt60Fe40/NG, suggesting that a Pt:Fe atomic ratio of ∼1:1 seems to be the optimum composition. The PtxFe100−x/NG catalysts show composition sensitive catalytic stability and the stability decreases in the sequence of Pt43Fe57/NG ≈ Pt60Fe40/NG > Pt27Fe73/NG > Pt79Fe21/NG > Pt20Fe80/NG > Pt16Fe84/NG ≈ 0. This is in accordance with the CV results.
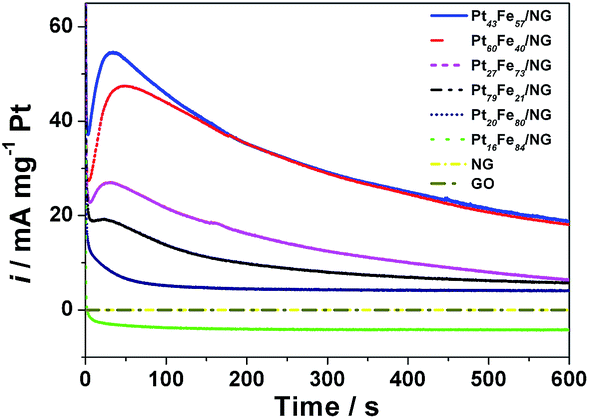 | ||
| Fig. 10 Chronoamperometric curves of the GO, NG, Pt/G and PtxFe100−x/NG catalysts in 0.5 M H2SO4 solution containing 1 M HCOOH. | ||
Conclusions
In summary, a series of PtxFe100−x/NG catalysts have been synthesized via a two-step strategy as catalysts for formic acid electrooxidation. The Pt:Fe ratio can be tuned freely and precisely. The doped nitrogen in the graphene supplies additional efficient anchoring sites for depositing metal particles uniformly. The electrochemical tests revealed that, alloying with Fe or using N-doped graphene as the support can evidently enhance the electrocatalytic performance of Pt. The composition sensitive catalytic performance of these catalysts was explored and the optimum composition in terms of the Pt:Fe atomic ratio was determined to be 1:1. On the basis of the synergistic interactions between the alloy catalysts and the NG substrate, the optimum Pt43Fe57/NG catalyst demonstrates amazingly improved electrocatalytic activity and stability with a reduced dosage of noble metal, making it a promising candidate for fuel cells applications. Moreover, the solutions developed in this manuscript will greatly benefit the rational design and preparation of high-performance metal catalysts, including Pt or Pd-based catalysts, for small organic molecule oxidation reactions and oxygen reduction reactions.
Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (21003079) and Shandong Provincial Natural Science Foundation, China (ZR2014JL015, ZR2014EMM004).Notes and references
- H. Zhang, M. Jin and Y. Xia, Chem. Soc. Rev., 2012, 41, 8035–8049 RSC.
- H. A. Grasteiger, D. R. Baker and R. N. Carter, in Hydrogen fuel cells: fundamentals and applications, Wiley-CPH, 2010 Search PubMed.
- I. E. L. Stephens, A. S. Bondarenko, U. Grønbjerg, J. Rossmeisl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 6744–6762 CAS.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- P. Yu, M. Pemberton and P. Plasse, J. Power Sources, 2005, 144, 11–20 CrossRef CAS PubMed.
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- J. Wang, R. M. Asmussen, B. Adams, D. F. Thomas and A. Chen, Chem. Mater., 2009, 21, 1716–1724 CrossRef CAS.
- J. Guo, Y. Sun, X. Zhang, L. Tang, H. Liu and H. Zhu, J. Alloys Compd., 2014, 604, 286–291 CrossRef CAS PubMed.
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- D. Xu, S. Bliznakov, Z. Liu, J. Fang and N. Dimitrov, Angew. Chem., Int. Ed., 2010, 49, 1282–1285 CrossRef CAS PubMed.
- W. Yu, M. D. Porosoff and J. G. Chen, Chem. Rev., 2012, 112, 5780–5817 CrossRef CAS PubMed.
- S. Guo and S. Sun, J. Am. Chem. Soc., 2012, 134, 2492–2495 CrossRef CAS PubMed.
- G. Wei, Y. Zhang, S. Steckbeck, Z. Su and Z. Li, J. Mater. Chem., 2012, 22, 17190–17195 RSC.
- Z. Ji, G. Zhu, X. Shen, H. Zhou, C. Wu and M. Wang, New J. Chem., 2012, 36, 1774–1780 RSC.
- T. Ghosh, Q. Zhou, J. M. Gregoire, R. B. van Dover and F. J. DiSalvo, J. Phys. Chem. C, 2010, 114, 12545–12553 CAS.
- B. Habibi and N. Delnavaz, RSC Adv., 2012, 2, 1609–1617 RSC.
- Y. Kang, L. Qi, M. Li, R. E. Diaz, D. Su, R. R. Adzic, E. Stach, J. Li and C. B. Murray, ACS Nano, 2012, 6, 2818–2825 CrossRef CAS PubMed.
- X. Zhao, J. Zhu, W. Cai, M. Xiao, L. Liang, C. Liu and W. Xing, RSC Adv., 2013, 3, 1763–1767 RSC.
- X. Xu, Y. Zhou, J. Lu, X. Tian, H. Zhu and J. Liu, Electrochim. Acta, 2014, 120, 439–451 CrossRef CAS PubMed.
- S. Zhao, H. Yin, L. Du, G. Yin, Z. Tang and S. Liu, J. Mater. Chem. A, 2014, 2, 3719–3724 CAS.
- G. Yang, Y. Li, R. K. Rana and J. J. Zhu, J. Mater. Chem. A, 2013, 1, 1754–1762 CAS.
- V. R. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross and N. M. Markovic, J. Am. Chem. Soc., 2006, 128, 8813–8819 CrossRef CAS PubMed.
- G. Krylova, N. M. Dimitrijevic, D. V. Talapin, J. R. Guest, H. Borchert, A. Lobo, T. Rajh and E. V. Shevchenko, J. Am. Chem. Soc., 2010, 132, 9102–9110 CrossRef CAS PubMed.
- N. G. Sahoo, Y. Pan, L. Li and S. H. Chan, Adv. Mater., 2012, 24, 4203–4210 CrossRef CAS PubMed.
- L. S. Panchakarla, K. S. Subrahmanyam, S. K. Saha, A. Govindaraj, H. R. Krishnamurthy, U. V. Waghmare and C. N. R. Rao, Adv. Mater., 2009, 21, 4726–4730 CAS.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 5, 11496–11500 CrossRef PubMed.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang and G. Yu, Nano Lett., 2009, 9, 1752–1758 CrossRef CAS PubMed.
- M. N. Groves, A. S. W. Chan, C. Malardier-Jugroot and M. Jugroot, Chem. Phys. Lett., 2009, 481, 214–219 CrossRef CAS PubMed.
- L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CAS.
- H. M. Jeong, J. W. Lee, W. H. Shin, Y. J. Choi, H. J. Shin, J. K. Kang and J. W. Choi, Nano Lett., 2011, 11, 2472–2477 CrossRef CAS PubMed.
- X. Wang, X. Cao, L. Bourgeois, H. Guan, S. Chen, Y. Zhong, D. M. Tang, H. Li, T. Zhai, L. Li, Y. Bando and D. Golberg, Adv. Funct. Mater., 2012, 22, 2682–2690 CrossRef CAS PubMed.
- L. Qu, Y. Liu, J. B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- R. I. Jafri, N. Rajalakshmi and S. Ramaprabhu, J. Mater. Chem., 2010, 20, 7114–7117 RSC.
- Y. Zhang, K. Fugane, T. Mori, L. Niu and J. Ye, J. Mater. Chem., 2012, 22, 6575–6580 RSC.
- C. Lamy, A. Lima, V. LeRhun, F. Delime, C. Coutanceau and J. M. Léger, J. Power Sources, 2002, 105, 283–296 CrossRef CAS.
- J. Willsau and J. Heitbaum, Electrochim. Acta, 1986, 31, 943–948 CrossRef CAS.
- Y. W. Rhee, S. Y. Ha and R. I. Masel, J. Power Sources, 2003, 117, 35–38 CrossRef CAS.
- L. S. Zhang, X. Q. Liang, W. G. Song and Z. Y. Wu, Phys. Chem. Chem. Phys., 2010, 12, 12055–12059 RSC.
- X. Xu, Y. Zhou, T. Yuan and Y. Li, Electrochim. Acta, 2013, 112, 587–595 CrossRef CAS PubMed.
- W. Chen, J. Kim, S. Sun and S. Chen, Langmuir, 2007, 23, 11303–11310 CrossRef CAS PubMed.
- Y. Kim, H. J. Kim, Y. S. Kim, S. M. Choi, M. H. Seo and W. B. Kim, J. Phys. Chem. C, 2012, 116, 18093–18100 CAS.
- J. Guo, B. Jiang, X. Zhang, X. Zhou and W. Hou, J. Solid State Chem., 2013, 205, 171–176 CrossRef CAS PubMed.
- G. Wang, J. Yang, J. Park, X. Gou, B. Wang, H. Liu and J. Yao, J. Phys. Chem. C, 2008, 112, 8192–8195 CAS.
- C. Xu, J. Sun and L. Gao, J. Mater. Chem., 2012, 22, 975–979 RSC.
- K. Zhang, Q. Yue, G. Chen, Y. Zhai, L. Wang, H. Wang, J. Zhao, J. Liu, J. Jia and H. Li, J. Phys. Chem. C, 2011, 115, 379–389 CAS.
- H. N. Dinh, X. Ren, F. H. Garzon, P. Zelenay and S. Gottesfeld, J. Electroanal.Chem., 2000, 491, 222–233 CrossRef CAS.
- Z. Lin, G. H. Waller, Y. Liu, M. Liu and C. P. Wong, Nano Energy, 2013, 2, 241–248 CrossRef CAS PubMed.
- R. B. Hye, S. Jin and S. H. Yang, Chem. Mater., 2011, 23, 3421–3428 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |