
Ligand–metal-drug coordination based micelles for efficient intracellular doxorubicin delivery†
Abstract
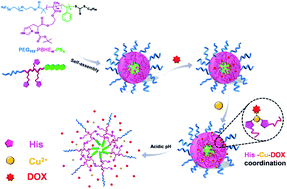
In the field of anticancer drug delivery, improving drug loading capacity of carriers and achieving efficient intracellular drug transportation simultaneously is very difficult but they are important issues for the development of chemotherapy. Herein, a reversible ligand–metal-drug coordination architecture responsive to pH was employed to construct a smart drug delivery system, in which the ligand and metal can be regarded as a harbor and an anchor to not only moor the drug (doxorubicin, DOX) but also send the cargo precisely on time. Based on the strategy, DOX loading content of the system could run up to 26.1%. Owing to the stability of coordination bonds at neutral conditions, premature drug leakage was extremely suppressed to lower than 5%, while an almost complete drug release was realized at pH 5.0 as a result of breakage of the bonds. Presence of the coordination interaction played a key role in controlled release of DOX, which followed the first-order kinetics model in the case of non-coordinated systems but the pseudo-second-order kinetics model in the case of coordinated systems. Moreover, the metal-coordinated system could effectively take DOX into HeLa cells, presenting a comparable cancer therapy effect to free DOX. This study established exploitation of coordination interaction to connect drugs and carriers as a promising way to meet urgent needs for chemotherapy.
 Please wait while we load your content...
Please wait while we load your content...

