
1-Ethyl-2,3-dimethylimidazolium paramagnetic ionic liquids with 3D magnetic ordering in its solid state: synthesis, structure and magneto-structural correlations†
Abel García-Saiza,
Imanol de Pedro*a,
Oriol Vallcorbab,
Pedro Migowskic,
Ignacio Hernándezad,
Luis Fernández Barquina,
Isaac Abrahamse,
Majid Motevallie,
Jairton Dupontc,
Jesús Antonio Gonzaleza and
Jesús Rodríguez Fernándeza
aCITIMAC, Facultad de Ciencias, Universidad de Cantabria, Santander 39005, Spain. E-mail: depedrovm@unican.es
bALBA Synchrotron Light Source, Cerdanyola del Vallés, Barcelona, Spain
cUniversidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
dMaterials Research Institute, School of Physics and Astronomy, Queen Mary University of London, London E1 4NS, UK
eMaterials Research Institute, Department of Chemistry and Biochemistry, School of Biological and Chemical Sciences, Queen Mary University of London, London E1 4NS, UK
First published on 24th June 2015
Abstract
Two novel paramagnetic ionic liquids, comprised of a 1-ethyl-2,3-dimethylimidazolium (Edimim) cation and a tetrahaloferrate(III) (FeX4) (X = Cl and Br) anion were synthetized and characterized by thermal, structural, Raman spectroscopy and magnetic studies. The crystal structures, determined by synchrotron X-ray powder diffraction and single crystal X-ray diffraction at 100 K for Edimim[FeCl4] and Edimim[FeBr4] respectively, are characterized by layers of cations (in non-planar configuration) and anions stacked upon one another in a three-dimensional (3D) manner with several non-covalent interactions: halide–halide, hydrogen bond and anion–π. Magnetization measurements show the presence of three-dimensional antiferromagnetic ordering below the Néel temperature (TN) with the existence of a noticeable magneto-crystalline anisotropy in the bromide compound. The corresponding magneto-structural correlations evidence that the 3D magnetic ordering mainly takes place via Fe–X⋯X–Fe (X = Cl and Br) interactions, displaying a higher superexchange magnetic interaction between the planes. Comparison with the Emim[FeX4] (X = Cl and Br) phases (Emim: 1-ethyl-3-methylimidazolium) reveals that the methylation at the C(2) position onto the imidazolium cation ring causes an increase of the melting point and a decrease of the TN. In contrast, the comparative study with Dimim[FeX4] (X = Cl and Br) compounds (Dimim: 1,3-dimethylimidazolium) shows a lower TN in the chloride compound, Edimim[FeCl4], whereas it is higher for the bromide, Edimim[FeBr4]. This fact is attributed to the spin delocalization of iron atoms in [FeBr4]− and discards the hypothesis that a bigger imidazolium ion size causes a weaker magnetic coupling in paramagnetic ionic liquids based on tetrahaloferrate anions and imidazolium cations with 3D magnetic ordering in its solid state.
Introduction
Paramagnetic ionic liquids are molten salts formed entirely of ions which have a melting point below 100 °C.1 They have received considerable attention among ionic liquids (ILs),2 fuelled by the possibility of tuning the materials properties by means of external magnetic fields3 or using them in the liquid state to produce nanoparticle-free magnetic emulsions and microemulsions.4 Moreover, they can combine magnetic5 and IL properties with additional intrinsic thermochromic,6 magneto-electrochromic7 or luminescent8 properties depending on the enclosed paramagnetic ion used. Thus, reports on the synthesis, study and application of these smart materials has proliferated. The first compounds developed since their discovery by the group of Hayashi,9 contained a metal anion (such as iron,10,11 copper,12 manganese13) and an organic cation, typically imidazolium, pyrrolidinium, pyridinium14 or tetraalkylphosphonium.15 Currently, the combinations of different rare-earth (europium,16 neodymium,17 dysprosium,5 etc.) chiral amino acids,18 dicationic ions19 tricationic ions20 or heteroanions,21 etc. have been studied for application in transport and separation of materials,22 separation of greenhouse gases (CO2) through supported magnetic ionic liquid membranes,23 magnetic surfactants,24 extraction of DNA25 or esterification of oleic acid to biodiesel.26ILs based on imidazolium cation have also recently received attention for the experimental27,28 and theoretical29 study of non-covalent interactions and cohesion properties.30,31 Convincing results indicate that the structural organization in the solid state can be extensible to the liquid phase and is apparently maintained to the gas phase.32,33 Many groups have focused their studies on the dipole–dipole, and hydrogen bonds within the crystal and molecular structures of these ILs34 in order to improve the understanding of these forces, aim at a fine tailoring of their technological applications. Importantly, in ILs based on imidazolium cation with paramagnetic anions, such as the presented compounds, it is also necessary to investigate other non-bonding interactions, like halogen–halogen35 (between the nearest metal complex anions) or anion–π36 (between the anion and cation) as these interactions play an important role in the organization of their structural units. In addition, if these non-bonding contacts are strong, interesting collective electrical and magnetic phenomena can arise, such as ferroelectricity in the bis(imidazolium) pentachloroantimonate(III), (C3N2H5)2SbCl5, or a three-dimensional (3D) magnetic ordering in ILs based on tetrahaloferrate ions.37,38 Moreover, changing the external conditions, such as pressure, shows an influence in the magnetic coupling, such as in Emim[FeCl4], which vary from antiferromagnetic (AF) to ferrimagnetic ordering.39
The learning of magneto-structural correlations in ILs based on tetrahaloferrate anion and imidazolium cation need a comprehensive understanding through a systematic investigation of a wide variety of compounds by changing the cationic and anionic structures. To accomplish this objective, we set out to study the family of ILs based on the tetrahaloferrate ion and 1,3-dimethylimidazolium cation. Our results reveal that (i) halogen–halogen interactions are the main force inducing the 3D magnetic ordering in the solid state of this type of ILs40 (ii) the spin population in the metal complex anion together with the distances and angles between the superexchange pathways [Fe–X⋯X–Fe (X = halide)] play a decisive role in attaining the 3D magnetic ordering40 and (iii) a less electronegative halide ion rises the efficiency of the magnetic couplings.41 Interestingly, it has also been shown that changes in the alkyl chain length of imidazolium cation cause variations in other physicochemical properties, including viscosity, density, conductivity, and melting point.42,43
Herein, we want to explore the effects on the thermal and magnetic properties of the methylation at C(2) position [N–C–N site] onto imidazolium cation, with different metal complex cations. We report on the synthesis, thermal, Raman and magnetic properties of two novel paramagnetic ionic liquids, namely 1-ethyl-2,3-dimethylimidazolium tetrachloroferrate, Edimim[FeCl4], and 1-ethyl-2,3-dimethylimidazolium tetrabromoferrate, Edimim[FeBr4], together with the crystal structure data with regard to obtaining the magneto-structural relationship.
Experimental section
Synthesis
All chemicals, anhydrous FeCl3 (99.9%, Aldrich), anhydrous FeBr3 (98.5%, Aldrich) 1,2-dimethylimidazole (98%, Sigma-Aldrich), acetonitrile (HPLC grade, Vetec), ethyl iodide (98%, Acros) were purchased from commercial sources and used without any further purification. All manipulations were made in air atmosphere, except otherwise stated in the text.1-Ethyl-2,3-dimethylimidazolium iodide
1,2-Dimethylimidazole (9.38 g, 97.5 mmol) was dissolved in 150 mL acetonitrile and ethyl iodide (11.68 g, 107.25 mmol) was added dropwise to the stirred solution cooled in an ice bath. After addition of the iodoethane, the reaction mixture was warmed up and the mixture refluxed for 24 h. The reaction was cooled down and the solvent removed under vacuum. The remaining solids were re-crystallized in a mixture of acetone: acetonitrile yielding white crystals 15.80 g, 79% yield.1-Ethyl-2,3-dimethylimidazolium tetrachloroferrate
1-Ethyl-2,3-dimethylimidazolium iodide (5 g, 19.84 mmol) was dissolved in 500 mL of distilled water and passed through an anion exchange column Amberlite IRA-400 column (OH-form) to yield a iodide free solution of 1-ethyl-2,3-dimethylimidazolium hydroxide (test by the Volhard method). The hydroxide solution was neutralized with concentrated hydrochloric acid (36%, Vetec) to pH 7 and water removed in rotary evaporator with the remaining water removed under vacuum at 100 °C. The solids were then dissolved in dichloromethane and dried over anhydrous Na2CO3, filtered and the solvent removed under vacuum, yielding 4.80 g (96%) of 1-ethyl-2,3-dimethylimidazolium chloride.In a glove-box, the 1-ethyl-2,3-dimethylimidazolium chloride (1 g, 4.90 mmol) was mixed with anhydrous FeCl3 (0.795 g, 4.90 mmol) in a 8 mL vial. The mixture was heated at 100 °C and the desired product was obtained as a red solid, 1.795 g (100% yield).
1-Ethyl-2,3-dimethylimidazolium tetrabromoferrate
1-Ethyl-2,3-dimethylimidazolium bromide was prepared the same way as 1-ethyl-2,3-dimethylimidazolium chloride, except for the use of hydrobromic acid (47%, Vetec) instead of HCl, affording 4.70 g (94% yield).In a glove-box, the 1-ethyl-2,3-dimethylimidazolium bromide (0.863 g, 4.23 mmol) was mixed with anhydrous FeBr3 (1.25 g, 4.23 mmol) in a 8 mL vial. The mixture was heated at 100 °C and the desired product was obtained as a brown-reddish solid, 2.113 g (100% yield).
Chemical analysis
Microanalytical data (C, H and N) were obtained with an Elemental model Vario 51 MACRO elemental analyzer. Iron, chlorine and bromine contents were determined with a Spectra Spectrometer DCP-AEC after dissolving a weighed amount of sample in water (aq). Chemical formula C5N2H9Cl4Fe and C5N2H9Br4Fe are confirmed. Thermal analysis: A Mettler-Toledo (TGA/SDTA851 and DSC822) was used for the thermal analyses in an oxygen dynamic atmosphere (50 mL min−1) at a heating rate of 10 °C min−1. For this experiment, ca. 15 mg of powder sample was thermally treated, and blank runs were performed.Elemental analysis
Elemental analysis for C7N2H13Cl4Fe (Edimim[FeCl4]). Calcd: C, 26.0; N, 8.67; H, 4.10; Cl, 43.93; Fe, 17.30%; found C, 25.8; N, 8.63; H, 4.16; Cl, 44.17; Fe, 17.24%. Elemental analysis for C7N2H13Br4Fe (Edimim[FeBr4]). Calcd: C, 16.79; N, 5.59; H, 2.63; Br, 63.84; Fe, 11.15%; found: 16.70; N, 5.54; H, 2.59; Br, 64.08; Fe, 11.09.Raman spectroscopy
The non-polarized Raman spectra were recorded in backscattering geometry with a Horiba T64000 triple spectrometer with a confocal microscope in subtractive mode that had a resolution of 0.6 cm−1. A 1800 grooves per mm grating and a 100 μm slit were used with a liquid N2-cooled CCD detector (Jobin-Yvon Symphony). A 647 nm line of a Coherent Innova Spectrum 70C Ar+–Kr+ laser was focused down with a 20× objective and the power on the sample kept below 5 mW to avoid laser-heating effects on the material being tested and the concomitant softening of the observed Raman peaks.Magnetic measurements
Temperature dependence of the magnetic susceptibility measurements were performed using a standard QD PPMS device by heating from 2 to 300 K at several magnetic fields, from 1 to 85 kOe, after cooling in either the presence (field cooling, FC) or the absence (zero field cooling, ZFC) of the applied field. Magnetization as a function of field (H) was measured using the same magnetometer in the −85 ≤ H/kOe ≤ 85 range at 2 and 15 K after cooling the sample in zero field. Heat capacity has been measured using the same magnetometer between 2 and 300 K at several magnetic fields, from 0 to 85 kOe, with a standard relaxation method using a two-tau model. In order to guarantee a good thermal contact, Apiezon N grease was used to glue the sample to the sample-holder. The addenda (sample-holder plus grease) was measured at different magnetic fields before the sample measurements and then subtracted from the total heat capacity in order to get the sample heat capacity. The sample was a 5 mg plate obtained compressing the original thin powder.Structural characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.44(1)), with atoms C1′, C1′′ and C2′′ common to both parts. H-atoms were refined separately for both parts in their geometrical positions using an atom-riding model. Anisotropic thermal parameters were refined for all non-hydrogen atoms, with those within the disordered imidazolium ring restrained to be similar to neighboring atoms. The final anisotropic full-matrix least-squares refinement on F2 with 146 variables and 72 restraints converged at R1 = 6.18%, for the observed data and wR2 = 15.63% for all data. The goodness-of-fit was 1.053. The largest peak and hole in the final difference electron density synthesis were 1.306 and −1.174 electrons per Å3, respectively, with an RMS deviation of 0.237 electrons per Å3. On the basis of the final model, the calculated density was 2.333 g cm−3, with F(000) = 470 electrons. Structure diagrams were generated within the Bruker Apex 2 suite of programs.
:0.44(1)), with atoms C1′, C1′′ and C2′′ common to both parts. H-atoms were refined separately for both parts in their geometrical positions using an atom-riding model. Anisotropic thermal parameters were refined for all non-hydrogen atoms, with those within the disordered imidazolium ring restrained to be similar to neighboring atoms. The final anisotropic full-matrix least-squares refinement on F2 with 146 variables and 72 restraints converged at R1 = 6.18%, for the observed data and wR2 = 15.63% for all data. The goodness-of-fit was 1.053. The largest peak and hole in the final difference electron density synthesis were 1.306 and −1.174 electrons per Å3, respectively, with an RMS deviation of 0.237 electrons per Å3. On the basis of the final model, the calculated density was 2.333 g cm−3, with F(000) = 470 electrons. Structure diagrams were generated within the Bruker Apex 2 suite of programs.Results and discussion
Thermal investigations show that Edimim[FeCl4] displays a solid–solid transition at 302 K and a melting point near 317 K. In Edimim[FeBr4], there is a solid–solid transition at 301 K and the melting point is located at 352 K (see Fig. 1). The melting point (Tm) of the bromide compound is markedly higher than that of chloride sample, consistent with greater interionic interactions present in Edimim[FeBr4]. A similar trend was observed for Emim [AlX4]50 (Tm: 281 K for X = Cl and 323 K for X = Br) and Emim [FeX4]51 (Tm: 291 K for X = Cl and 316 K for X = Br) ILs. Upon further heating, Edimim[FeCl4] starts to decompose at a temperature of 548 K and Edimim[FeBr4] at 571 K, offering a wide liquid region ≈ 200 K (see inset of Fig. 1). | ||
| Fig. 1 DSC-thermogram of (a) Edimim[FeCl4] and (b) Edimim[FeBr4]. Black line: 1st cooling cycle; blue line: 2nd heating cycle. Heating rate 10 K min−1. The insets show the TGA curves of Edimim[FeCl4] and Edimim[FeBr4], respectively recorded in inert atmosphere. | ||
The crystal structure of Edimim[FeCl4] at 100 K was solved from synchrotron X-ray powder diffraction data using the direct-space method TALP46 and refined with the restrained Rietveld refinement program RIBOLS using distance restraints taken from Mogul.47 Hydrogen atoms were placed in calculated positions and constrained to the corresponding C atoms in the final set of refinement cycles. The experimental, calculated and difference powder profiles are displayed in Fig. S1.† For Edimim[FeBr4], the crystal structure was determined by single crystal X-ray diffraction at 100 K. Atomic coordinates, interatomic distances and thermal parameters for both compounds are listed in the ESI† as well as the corresponding CIF files. Crystallographic data and structure refinement details are presented in Table 1. The most relevant intermolecular distances are displayed in Tables 2 and 3.
| Molecular formula | C7H13Cl4FeN2 | C7H13Br4FeN2 |
| Formula weight | 322.84 | 500.68 |
| Temperature (K) | 100(1) | 100(2) |
| Wavelength (Å) | 0.6193 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21 |
| Unit cell dimensions | a = 9.6703 (1) Å α = 90° | a = 6.957(6) Å α = 90° |
| b = 14.3513 (2) Å β = 94.261(1)° | b = 14.804(12) Å β = 90.728(14)° | |
| c = 9.5744 (1) Å γ = 90° | c = 7.232(6) Å γ = 90° | |
| Volume (Å3) | 1325.08(3) | 744.8(11) |
| Z | 4 | 2 |
| Density (calculated) (g cm−3) | 1.618 | 2.233 |
| Measured 2θ range, stepsize (°) | 1.104–43.092, 0.006 | |
| Rietveld refinement details: | ||
| Profile function | Pseudo-Voigt | |
| 2θ range used | 4.002–39.996 | |
| Num. of reflections | 2330 | 4861 |
| Data points | 4999 | |
| Parameters | 54 | |
| Restraints | 36 | |
| Rwp | 0.060 | |
| χRietveld | 6.130 | |
| χRietveld/χPattern-matching | 1.692 | |
| F(000) | 470 | |
| Crystal size | 0.10 × 0.06 × 0.06 mm3 | |
| Theta range for data collection | 2.75 to 28.38° | |
| Index ranges | −9 ≤ h ≤ 9, −19 ≤ k ≤ 18, −5 ≤ l ≤ 9 | |
| Independent reflections | 3158 [R(int) = 0.0581] | |
| Completeness to theta = 28.38° | 95.0% | |
| Max. and min. transmission | 0.3963 and 0.7457 | |
| Refinement method | Full-matrix least-squares on F | |
| Data/restraints/parameters | 3158/72/146 | |
| Goodness-of-fit on F2 | 1.053 | |
| Final R indices [I > 2sigma(I)] | R1 = 0.0618, wR2 = 0.1371 | |
| R indices (all data) | R1 = 0.1135, wR2 = 0.1563 | |
| Largest diff. peak and hole | 1.306 and −1.174 Å−3 |
| Edimim[FeCl4] at 100 K | ||
|---|---|---|
| Length (Å) | Angle (°) | |
| a Distance smaller than the sum of vdW radii (3.05 Å for C–H⋯Cl).b Angle between Fe–centroid vector and the imidazolium ring plane.c Sum of vdW radii for Cl⋯Cl is 3.7 Å. | ||
| H⋯Cl (potential hydrogen bonds)a | ||
| C1′–H1A′⋯Cl2 | 3.011(10) | 139.1(8) |
| C1′–H1B′⋯Cl3 | 2.981(6) | 153.7(8) |
| C1′′–H1A′′⋯Cl3 | 3.040(11) | 124.1(9) |
| C1′′–H1C′′⋯Cl1 | 3.017(11) | 141.2(7) |
| C1′′′–H1B′′′⋯Cl1 | 2.912(12) | 148.4(5) |
| C1′′′–H1A′′′⋯Cl1 | 2.995(11) | 165.3(5) |
| C1′′′–H1C′′′⋯Cl4 | 3.149(9) | 120.7(9) |
| C2′–H2B′⋯Cl4 | 3.041(11) | 116.5(8) |
| C2′–H2B′⋯Cl2 | 2.993(7) | 122.7(4) |
| C4–H4⋯Cl1 | 2.846(11) | 154.7(8) |
| C4–H4⋯Cl4 | 2.996(6) | 117.3(7) |
| C5–H5⋯Cl3 | 2.751(10) | 148.7(10) |
| C5–H5⋯Cl4 | 2.976(9) | 117.8(10) |
|
||
| [FeCl]−⋯[Dimim]+ (potential π–d interactions) | ||
| Fe⋯centroid | 4.425(5) | 70.8b |
| Cl4⋯N3 | 3.401(10) | |
| Cl4⋯C2 | 3.548(11) | |
| Cl2⋯C4 | 3.388(12) | |
| Cl2⋯N3 | 3.705(9) | |
| Cl4⋯[C4–N3] | 3.414(10) | |
| Cl2⋯[C4–N3] | 3.481(10) | |
| Cl2⋯centroid | 3.714(6) | 101.01(16) |
| Cl4⋯centroid | 3.454(6) | 100.43(16) |
|
||
| Cl⋯Cl interactionsc | ||
| Cl3⋯Cl4 | 4.171(5) | |
| Cl3⋯Cl1 | 3.936(5) | |
| Cl3⋯Cl2 | 4.025(5) | |
| Cl2⋯Cl1 | 4.192(4) | |
| Edimim[FeBr4] at 100 K | ||
|---|---|---|
| Length (Å) | Angle (°) | |
| a Distance smaller than the sum of vdW radii (3.15 Å for C–H⋯Br).b Angle between Fe–centroid vector and the imidazolium ring plane.c Sum of vdW radii for Br⋯Br is 3.9 Å. | ||
| H⋯Br (potential hydrogen bonds)a | ||
| C1′′′–H1B′′′⋯Br4 | 2.725(6) | 145.9(4) |
| C1′′′–H1B′′′⋯Br1 | 3.089(7) | 119.5(6) |
| C5–H5⋯Br2 | 3.105(6) | 156.1(11) |
| C4–H4⋯Br1 | 2.790(10) | 161.8(7) |
| C1′′–H1A′′⋯Br4 | 3.076(4) | 136.3(6) |
| C1′′–H1B⋯Br1 | 3.059(6) | 144.2(9) |
| C1′′–H1B′′⋯Br3 | 3.133(5) | 132.3(10) |
|
||
| [FeBr]−⋯[Dimim]+ (potential π–d interactions) | ||
| Fe⋯centroid | 4.601(8) | 67.4b |
| Br3⋯C2 | 3.748(6) | |
| Br3⋯N3 | 3.734(12) | |
| Br3⋯[N1–C2]c | 4.001(6) | |
| Br3⋯[N3–C2]c | 3.676(13) | |
| Br3⋯centroid | 4.016(10) | 61.4(14) |
| Br4⋯centroid | 4.236(9) | 60.2(12) |
|
||
| Br⋯Br interactionsc | ||
| Br3⋯Br4 | 4.095(4) | |
| Br4⋯Br1 | 3.985(5) | |
| Br3⋯Br2 | 4.053(6) | |
Fig. 2 illustrates the crystal structure of Edimim[FeCl4] and Edimim[FeBr4] at 100 K. Edimim[FeCl4] crystallizes in the monoclinic crystal system, space group P21/n, with a = 9.6703(1), b = 14.3513(2), c = 9.5744(1) Å and β = 94.261(1)° [V = 1325.08(3) Å3, Z = 4, ρcalcd = 1.618 g cm−3]. The crystal structure of Edimim[FeBr4] is also monoclinic, space group P21, a = 6.957 (6) Å, b = 14.804(12) Å, c = 7.232(6) Å and β = 90.728(14)° [Z = 2, ρcalcd = 2.23 g cm−3]. Both frameworks, projected along b direction, can be described as a stacking of [Edimim]+ and [FeX4]− (X = Cl and Br) intercalated layers changing orientation from layer to layer, with Fe⋯Fe distances larger than 6.5 Å inside the layer (Fig. S2†). For Edimim[FeCl4] they also lie antiparallel to each other along the c axis, while they are stacked identically one above the other in the a direction. The topological description of the [FeX4]− units (X = Cl and Br) of both compounds involves a tetrahedral geometry, with a mean X–Fe–X bond angle of 109.5(2) and 109(1)° and mean Fe–X bond distances of 2.21(1) and 2.36(1) Å for chloride and bromide derivatives, respectively. In the case of [Edimim]+ units, the imidazole ring is aromatic and planar with all the refined values for the C–C and C–N bond lengths laying in the expected range of other imidazolium compounds.41,52 Furthermore, the conformational equilibria of the imidazolium cation has a gauche (non-planar) conformation with respect to NCC angle of the ethyl group, in good agreement with the conformation found in other ILs with 1-ethyl-3-methylimidazolium cation.39,53 The values of the C2–N1–C1′–C2′ torsion angle for Edimim[FeCl4] point to 85(1)°, whereas for Edimim[FeBr4] the corresponding one is 83(3)°.
 | ||
| Fig. 2 Crystal packing in the bc plane of (a) Edimim[FeCl4] and (b) Edimim[FeBr4] crystal structures at 100 K. Orange: iron, green: chloride, gold: bromine, grey: carbon and blue: nitrogen. H atoms are omitted for clarity. | ||
Inspection of the crystallographic data of both compounds at 100 K and corresponding contrast with each specification rule, three types of non-covalent interactions are detected: (a) halide–halide, (b) hydrogen bonds and (c) anion–π. Four halide–halide interactions between the nearest [FeCl4]− metal complex anions (Fig. 3a) arise in Edimim[FeCl4]. Similarly, [FeBr4]− has three probable contacts in the unit cell (Fig. 3b), in the case of Edimim[FeBr4]. These non-covalent interactions propagate in a zigzag manner along the b axis [4.171(5) and 4.095(4) Å] and as linear chains across ac plane [distances between 3.936(5) and 4.192(4) Å and from 3.985(5) to 4.053(6) Å for Cl and Br, respectively]. These interactions must be taken into consideration even though they are longer than the sum of the vdW-radii (<3.7 and 3.9 Å, respectively), since they are responsible for establishing of the 3D magnetic ordering (see the magnetic results below). The Fe⋯Fe distances are too long to consider direct magnetic interaction between metal atoms. They are in agreement with previous studies about the most frequent stacking distance of two metal complex anion in other paramagnetic ILs based on halometallates.40,54 Cation–anion interactions are characterized by an anisotropic H bonding network of twelve and nine hydrogen bonds between the halides and the surrounding Edimim+ cations for Edimim[FeCl4] and Edimim[FeBr4], respectively (according to IUPAC recommendation for H bonds; Tables 2 and 3 and Fig. 4). The shortest H-bond is between C5–H5⋯Cl3 and C1′′-H′1B′′′⋯Br4, with hydrogen contact distances of 2.751(1) and 2.725(1) Å, respectively. It is worth noting that the nearest imidazolium centroid⋯Cl− contact for Edimim[FeCl4] [3.454(6) and θ = 100.43(16)°] is smaller than the most common value considered to indicate anion–π interaction (≤3.65 Å and θ = 90 ± 10°).55 However, the corresponding distance for Edimim[FeBr4] [4.016(10) and θ = 61.4(14)°] is larger than the specification rule. Nevertheless the Edimim[FeBr4] compound displays two contacts above the ring periphery which are smaller than the sum of the vdW radii in Br⋯C and Br⋯[C–N] (≤3.75 and 3.70 Å respectively). These show interaction distances of 3.734(12) and 3.678(6) Å for Br3⋯C2 and Br3⋯[N3–C2] respectively [see Fig. 5 and Tables 2 and 3], representative of weak anion–π interactions.56
 | ||
| Fig. 3 Schematic view of the halide–halide interactions (dashed lines; possible magnetic exchange pathways) for (a) Edimim[FeCl4] via Fe–Cl⋯Cl–Fe bridges and (b) Edimim[FeBr4] via Fe–Br⋯Br–Fe bridges. Distance units in Å. | ||
 | ||
| Fig. 4 Hydrogen-bonding network in (a) Edimim[FeCl4] and (b) Edimim[FeBr4]. H–X (X = Cl and Br) contacts up to 3.05 and 3.15 Å are plotted with dashed lines. Distance units in Å. | ||
 | ||
| Fig. 5 Schematic view of the cation and anion arrangement (dashed lines) within the main contact distances for (a) Edimim[FeCl4] and (b) Edimim[FeBr4]. Distance units in Å. | ||
Fig. 6 shows the non-polarized Raman spectrum of the Edimim[FeCl4] sample between 50 and 3300 cm−1 at room temperature with excitation at 647 nm. These data offer the possibility of studying the conformational analysis of the Edimim+ cation and the metal complex anion of this phase at room temperature (RT). The measured frequencies and their corresponding assignment in terms of the involved vibrational modes of the molecular ions are summarized in Table 4. We observe four Raman-active modes in the low-frequency region (see left-inset of Fig. 6, black) belonging to the symmetry point group Td of the [FeCl4]− iron complex. The peaks at νs = 110 and νas = 135 cm−1 are attributed to the bending modes and the other two, νs = 332 and νas = 338 cm−1, respectively are related to the stretching modes of the Fe–Cl bond. In the same inset of Fig. 6 also shown are the four Raman-active vibration modes of the [FeBr4]− iron complex of Edimim[FeBr4] at RT (in red), with frequencies νs = 70 and νas = 90 cm−1 (bending modes) and νs = 201 and νas = 285 cm−1 (stretching). All these vibrational data agree with literature values for other compounds with tetrahaloferrate(III) anions,57,58 corroborating that the presented compounds encompass the [FeX4]− (X = Cl and Br) units, ruling the presence of [Fe2X7]− dimers.
 | ||
| Fig. 6 Raman spectra of Edimim[FeCl4] between 100 and 3300 cm−1 at 300 K. The left inset shows the low energy region of Raman spectra of Edimim[FeCl4] and Edimim[FeBr4] at 300 K. The right inset shows the high energy region of Raman spectra of Edimim[FeCl4] at 300 K. | ||
| Experimental center/cm−1 | Assignation |
|---|---|
| 110 | [FeCl4]− Fe–Cl sym bend |
| 135 | [FeCl4]− Fe–Cl asym bend |
| 332 | [FeCl4]− Fe–Cl sym stretch |
| 338 | [FeCl4]− Fe–Cl asym stretch |
| 394 | [Ethyl] CH2–N bend, gauche conformer |
| 579 | [Ring] ip sym bend, [ethyl] CH2–N stretch, [ring CH3] CH3–N stretch |
| 665 | [Ring] ip asym bend, [ethyl] CH2–N stretch, [ring CH3] CH3–N stretch |
| 711 | [Ring] ip asym bend, [ring CH3] CH3–N stretch |
| 725 | [Ring] HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH sym bend, [ethyl] CH2–N stretch, [ring CH3] CH3–N stretch CH sym bend, [ethyl] CH2–N stretch, [ring CH3] CH3–N stretch |
| 774 | [Ring] C–C sym stretch, [ring] HCCH sym bend, [Ring] N–C(H)–N op sym bend |
| 801 | [Ring] HCCH sym bend, [ring] NC(H)N op bending, |
| 958 | [Ring] ip sym bend, [ethyl] C–C stretch |
| 1088 | [Ring] ip sym stretch, [ethyl] CH2–N stretch, [ring CH3] CH3–N stretch |
| 1335 | |
| 1381 | [Ring] ip asym stretch, [ethyl] C–C stretch, [ethyl] CH2–N stretch, [terminal CH3] CH3–N stretch |
| 1421 | [Ring] ip asym stretch, [ethyl] C–C stretch, [ethyl] CH2–N stretch, [ring CH3] CH3–N stretch |
| 1452 | [Ring] ip asym stretch, [ethyl] C–C stretch, [ethyl] CH2–N stretch, [ring CH3] CH3–N stretch |
| 2831 | [Terminal CH3] H–C–H sym stretch |
| 2879 | [Ring CH3] H–C–H sym stretch |
| 2932 | [Ethyl] H–C–H sym stretch |
| 2965 | [Ethyl] H–C–H asym stretch |
| 3000 | [Ethyl] H–C–H asym stretch |
| 3079 | [Terminal CH3] H–C–H asym stretch |
| 3101 | [Ring CH3] H–C–H asym stretch |
| 3130 | [Ring N–C(H)–N] C–H stretch |
| 3149 | [Ring] HCCH sym stretch. [Ring] ip sym stretch |
| 3174 | [Ring] HCCH sym stretch. [Ring] ip sym stretch |
The Raman bands observed between 390 and 3200 cm−1 (Fig. 6 and Table 4) of Edimim[FeCl4] are assigned to the Edimim+ cation. These bands may be compared with the theoretical and experimental vibrational frequencies of Emim+39 and Dimim+ cation;40 which have been previously reported for other members of the imidazolium-based paramagnetic ILs. We attribute the Raman peak located at 394 cm−1 to the gauche non-planar geometry of the conformers53,59 given the conformational equilibrium of the Emim+ cation across the CH2–N bending mode of the ethyl chain [(trans–gauche) (planar–non-planar)]. This peak is also observed in the bromide compound at 303 cm−1. The absence of a Raman signal between 430 and 450 cm−1 (where the vibrational frequencies of the trans planar geometry typically appear) rules out the presence of this conformer. The ring in-plane symmetric stretching and bending modes60 (see Table 4) are found in the 500–1600 cm−1 range. Finally, the most intense peaks between 2700 and 3200 cm−1 (see right-inset of Fig. 6 and Table 4) correspond to the terminal CH3, the ring CH3 and the ethyl chains (symmetric and asymmetric H–C–H stretch) and the other, weaker ones (some of which overlap), to a ring N–C(H)–N (C–H stretch) modes, and a mixture of a ring HCCH symmetric stretch and a ring in-plane symmetric stretch modes.39,60
Variable temperature magnetic susceptibility measurements of the presented compounds were carried out on powdered samples in the 2–300 K temperature range. The temperature dependence of the molar magnetic, χm, and the reciprocal, χm−1, susceptibilities measured under 1 kOe are represented in Fig. 7. The linear behavior of χm−1 at temperatures higher than 20 K can be fitted to a Curie–Weiss law for both compounds. The calculated values give an effective paramagnetic moment (μeff) of 5.62 and 5.93 μB and paramagnetic Weiss temperatures (θP) of −1.0 and −12.5 K for Edimim[FeCl4] and Edimim[FeBr4], respectively. Both effective paramagnetic moments agree with the expected value for high spin d5 Fe(III) ions (μeff = 5.92 μB per Fe ion) and the negative θP is a signature of an overall antiferromagnetic (AF) interactions when both compounds are frozen, in good agreement with those found in other paramagnetic ILs40,41 and paramagnetic salts61 based on tetrahaloferrate ions. In addition, the results confirm that a less electronegative halide ion in the metal complex cation rises the efficiency of the magnetic couplings.41
 | ||
| Fig. 7 Temperature dependence of the molar susceptibility (χm) and 1/χm measured at 1 kOe for (a) Edimim[FeCl4]; (b) Edimim[FeBr4]. The solid red lines are the fit according to Curie–Weiss law. The insets show an enlargement of the low temperature region of (χm). | ||
In the low temperatures regime, χm increases and reaches a maximum at 2.9 K and 9 K for Edimim[FeCl4] and Edimim[FeBr4], respectively, (see the insets of Fig. 7) denoting the existence of long-range magnetic order, with a gradual decrease in the susceptibility of ca. 15%, which is usual for non-oriented antiferromagnets. It is worth mentioning that the replacement of Cl with Br in Edimim[FeX4] leads to an increase in the Néel temperature (TN), favoring stronger magnetic interactions. This agrees with findings in Emim[FeX4] (TN: 4.2 K for X = Cl and 12.5 K for X = Br)38,51 and Dimim[FeX4] (TN: 5.6 K for X = Cl and 7.7 K for X = Br)40,41 paramagnetic ILs. However, the Néel temperature of the bromide compound is higher than that obtained for Dimim[FeBr4]. This fact goes against our first hypothesis of a smaller chain length of the organic cation causing an increase of the efficiency in the transmission of the magnetic interactions, resulting in an increase in the ordering temperatures in paramagnetic ILs based on tetrahaloferrate ions. On the other hand, χm maxima shift to lower temperatures with increasing magnetic field (see Fig. 8), as expected for an AF ordering, and disappear for larger magnetic fields (60 kOe) for Edimim[FeCl4]. However, for Edimim[FeBr4] the maximum in susceptibility does not disappear in the 0–80 kOe range, this field not being strong enough to break the AF ordering. This is an evidence of stronger magnetic couplings, as observed in Dimim[FeBr4].41
 | ||
| Fig. 8 Low temperature ZFC magnetic susceptibility at different applied magnetic fields for (a) Edimim[FeCl4]; (b) Edimim[FeBr4]. | ||
Fig. 9 depicts the magnetization as a function of the magnetic field (H) at 2 K for Edimim[FeX4] (X = Cl and Br). The magnetization shows no hysteresis at this temperature, in the ordered state (see inset of Fig. 9), thereby excluding the existence of any ferromagnetic component. Moreover, in the case of Edimim[FeCl4] the magnetization tends to saturate above 40 kOe with a value of ≈4.6 μB per Fe ion, which is near the expected fully-saturated value of 5 μB per Fe for Fe(III) ion. However, for Edimim[FeBr4] the magnetization rises continuously, with an small inflexion near 30 kOe, and does not show saturation up to 85 kOe. In fact, the magnetization value attained in this magnetic field (≈2.30 μB per Fe ion) is far from the expected fully-saturated value, which confirms a stronger magnetocrystalline anisotropy within this material with respect to the chloride analogue, similar to other analogous paramagnetic ILs based on tetrahaloferrate with 3D ordering.11,40,41 The M(H) curves measured between 2 K and the paramagnetic state (see Fig. S3†) allowed us to prepare the Arrott plots (M2 vs. H/M). A change of regime below 3.0 and 9 K is found in the low temperature region for the chloride and bromide compounds respectively, indicating the appearance of long-range AF order in agreement with the magnetic susceptibility measurements.
 | ||
| Fig. 9 Magnetization vs. applied magnetic field for Edimim[FeCl4] and Edimim[FeBr4] at 2 K. | ||
Fig. 10 shows the temperature dependence of the molar heat capacity (CP) for the presented compounds, between 2 and 300 K. In the absence of an external magnetic field, CP reveals a maximum (ΔCp = 19 and 8 J mol−1 K−1) at 2.9 and 8.8 K for Edimim[FeCl4] and Edimim[FeBr4], respectively (see upper insets in Fig. 10). This anomaly reveals a second-order transition and it can be related to the establishment of 3D antiferromagnetic order. The heat capacity measurements in the presence of an applied magnetic field show that the maxima get smaller and shift toward lower temperatures as the field increases. In addition, the magnetic peak of the bromide compound does not disappear for fields larger than 80 kOe, corroborating the presence of the stronger magnetocrystalline anisotropy, as detected in the magnetic susceptibility data.
 | ||
| Fig. 10 Specific heat of (a) Edimim[FeCl4] and (b) Edimim[FeBr4] between 2 and 300 K. Lower inset shows the experimental data (black full dots), estimated phonon contribution (red dashed line) and magnetic contribution (blue full dots). The upper inset is the specific heat at different applied magnetic field (H ≤ 85 kOe). | ||
Cp increases continuously above the magnetic peak and below 280 K in both compounds due to the phonon contribution. The curves show an inflexion point for T > 280 K, associated with the beginning of the solid–solid transition detected in the calorimetric measurements, (see Fig. 1) near RT. Interestingly, the value at 280 K (≈330 J mol−1 K−1) it is still far from the value derived from the Dulong–Petit law (Cp = 675 J mol−1 K−1 at RT) due to the presence of a high number of hydrogen atoms within the imidazolium cation, displaying high excitation energies.62,63
The magnetic contribution to the heat capacity (Cmag) of both phases was determined by subtracting the phonon contribution (Cpho) up to 270 K, considering a Debye model64 with the existence of three Debye temperatures (θ).41 The highest temperature (θ1) relates to the n1 lighter atoms (hydrogen atoms), θ2 and n2 corresponds to the medium-weight atoms (C and N), and θ3 and n3 to heavier atoms (Fe, Cl or Br); with n1+ n2+ n3 = N being N the number of the atoms in the unit cell. The thermal variation of Cmag is presented in the lower insets of Fig. 10. The best fit for the experimental data of Edimim[FeCl4] at temperatures higher than 15 K yields θ1 = 1840 K, θ2 = 315 K, θ3 = 99 K, n1 = 17.7, n2 = 5.7 and n3 = 3.6; similar parameters that was obtained for Edimim[FeBr4]. We should point out that although it is a simplified model, it allows to estimate reasonable well the non-magnetic contribution in the low temperature region (T < 20 K) (see low insets of Fig. 10). As can be see, Cmag progressively increases for Edimim[FeBr4] as the temperature becomes smaller until it reaches the maximum at the onset of the 3D magnetic ordering. Moreover, in Edimim[FeCl4], a shoulder around 10 K suggests low-dimensional ordering65 or short-range magnetic ordering,66 prior to the establishment of the 3D magnetic ordering. The integral of Cmag/T with respect to the temperature, provides the magnetic entropy (see Fig. S4†). Both samples show a value near 14.8 J mol−1 K−1 corresponding with the full entropy ΔS = Rln((2 × 5/2) + 1) (R = 8.31 J mol−1 K−1) for a system with high spin d5 Fe(III) ions, spin S = 5/2.
The role of the organic cation in the efficiency of the transmission of the magnetic interactions of paramagnetic ILs can be studied from the analysis of the magnetostructural correlations of the presented compounds and the comparison with the paramagnetic ionic liquid analogues, Dimim[FeCl4] and Dimim[FeBr4], which we have studied in a previous work. The crystal structures of Edimim[FeCl4] and Edimim[FeBr4] at 100 K give no evidence for direct iron–iron interactions. At this temperature, the Fe⋯Fe distances are 6.547 and 7.053 Å inside a layer (in the a–c plane, respectively) and 8.345 Å in the b direction [see Fig. S2(a)† and Table 2]. For bromide compound, the iron–iron distances are 6.957 and 7.232 (in plane) and 8.667 Å (along b), respectively [see Fig. S2(b)† and Table 3]. Therefore, the 3D antiferromagnetic ordering detected by magnetic measurements must take place via super-exchange coupling, the Fe–X⋯X–Fe (X = Cl and Br) interaction being the main exchange pathway. In addition, the interactions distances between halide and imidazolium (Fig. 5) along the b direction promote a weak orbital coupling between Fe3+ ions. Thus, indirect exchange coupling of the type Fe–X⋯Im⋯X–Fe, which should be weakest due to the involvement of an imidazolium donor cation (defined as S = 1/2)67 inside the magnetic exchange-pathway could be present in these compounds. Moreover, it would seem reasonable to suggest that if the crystal structure of both compounds at 100 K is maintained below TN, three direct superexchange pathways are detected: (i) one magnetic J interplanar interaction (J1 or J⊥), which gives rise to zig-zag chains forming a ladder structure that runs parallel to the b direction (ii) two intraplane couplings, J2 and J3 (J∥), which link the iron atoms in linear ferromagnetic chains in the ac plane (Table 5). Despite the fact that X⋯X distances are slightly longer than the sum of the vdW-radii of two halide atoms (see Fig. 3), the data show a remarkable agreement with the contact distances reported in other paramagnetic ILs44 and metal–organic materials35,68 with 3D magnetic ordering that show this type of magnetic coupling. In addition, a decrease of these distances is expected below TN, due to thermal effects, and hence we cannot rule out indirect superexchange anion–anion interactions (Fe–X⋯Im⋯X–Fe).41 This mechanism has been proposed for the Dimim FeBr4, where the magnetic structure was determined.
| Magnetic exchange pathways | Direct distance Fe–Fe/Å | Length of exchange pathway/Å | Bond/Å Fe–X | Bond/Å X–X | Bond/Å X–Fe | Angle/° Fe–X–X | Angle/° X–X–Fe | Torsion angle (τ)/° | Direction |
|---|---|---|---|---|---|---|---|---|---|
| Dimim[FeCl4] | |||||||||
| J1 (exp.) | 8.345(2) | 8.578 | 2.211 | 4.171 | 2.196 | 168.90 | 152.65 | 70.38 | b |
| J2 (exp.) | 6.547(3) | 8.632 | 2.229 | 4.025 | 2.211 | 143.53 | 82.14 | 168.57 | a–c |
| J2 (exp.) | 6.547(3) | 8.632 | 2.229 | 4.192 | 2.211 | 146.48 | 78.55 | 165.64 | a–c |
| J3 (exp.) | 7.053(2) | 8.386 | 2.229 | 3.936 | 2.221 | 108.18 | 138.21 | 125.29 | a–c |
|
|||||||||
| Dimim[FeBr4] | |||||||||
| J1 (exp.) | 8.667(3) | 8.812 | 2.363 | 4.095 | 2.353 | 166.85 | 161.17 | 84.34 | b |
| J2 (exp.) | 6.957(4) | 8.691 | 2.355 | 3.985 | 2.351 | 158.02 | 91.19 | 34.15 | a–c |
| J3 (exp.) | 7.232(2) | 8.776 | 2.372 | 4.053 | 2.351 | 151.28 | 102.28 | 71.72 | a–c |
The strength of the magnetic exchange pathways inside both compounds can be discussed and compared observing the following parameters: (i) X⋯X distance, (ii) Fe–X⋯X–Fe angles and (iii) the Fe–X⋯X–Fe torsion angle (τ) (Table 5). Comparing the X⋯X distances, the interplanar values (J1) (4.171 and 4.095 Å for Cl and Br, respectively) are larger than the intraplane interactions (J2 and J3) [4.025 (lowest distance of J2) and 3.936 for Cl and 3.985 and 4.053 Å for Br]. For (ii) and (iii) parameters, the intraplane interactions (J2 and J3) show a mixture of exchange angles: Fe–X⋯X, between 138.21 and 158.02° and X⋯X–Fe, from 78.55° and 108.18°, with τ ranging from 34.15 to 168.57°. However, J1 displays a nearly linear combination of Fe–X⋯X and X⋯X–Fe angles (from 168.90 to 152.65°) with τ = 70.38 and 84.34° for Cl and Br, respectively. Thus, the interplanar magnetic interaction is found to be stronger than the intraplanar ones in both compounds, according to the rule that shorter halide–halide distances, larger Fe–X⋯X–Fe angles, and Fe–X⋯X–Fe torsion angles near 90° correspond to stronger magnetic exchange constants;35 which agrees with the reports on Dimim[FeCl4] and Dimim[FeBr4]41 analogues.
The exchange couplings between the Fe3+ ions can also be quantitatively analyzed from the macroscopic experimental parameters. The best model to describe the magnetic susceptibility data includes a combination of a modified expression of the classical spin ladder-like chain (1D) for interplanar interactions together with a mean-field term with spin 5/2, to account for the intraplanar ones (2D).69
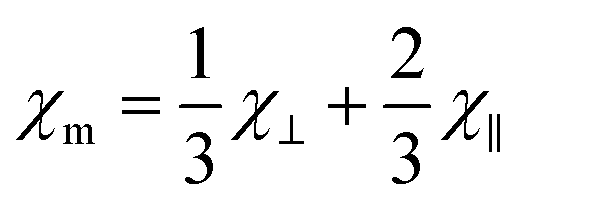
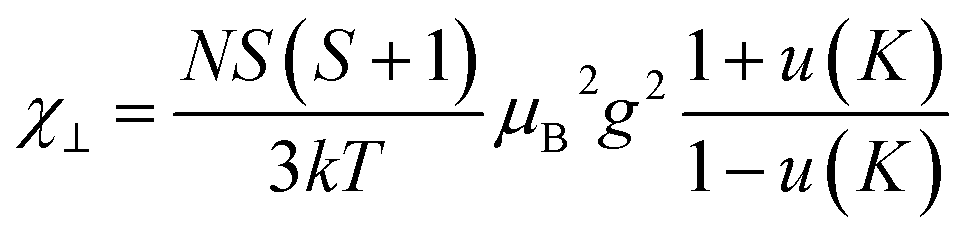 | (1) |
| u(K) = cothK − 1/K |
| K = −2J⊥S(S + 1)/kT |
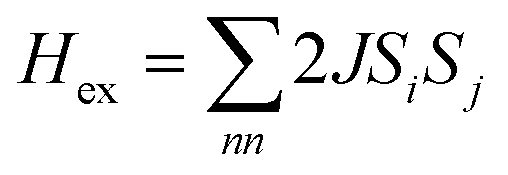 . In addition, the magnetic susceptibility for the 2D AF framework, χ∥ for S = 5/2 is given by the Rushbrooke and Wood expression72
. In addition, the magnetic susceptibility for the 2D AF framework, χ∥ for S = 5/2 is given by the Rushbrooke and Wood expression72
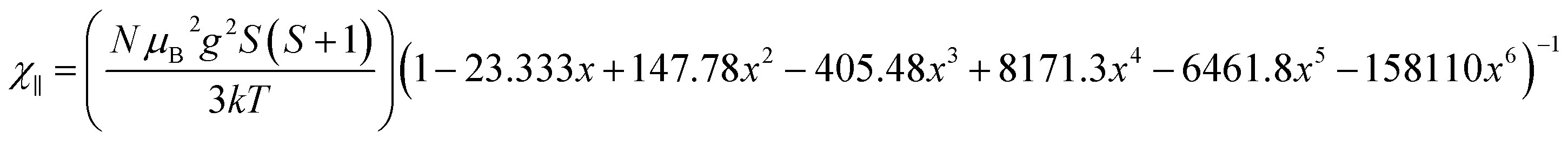 | (2) |
The least-squares fit of the experimental data from 2 to 300 K [solid line in Fig. 11], yields g = 2.00 and 2.006, J⊥ = −0.050 and −0.195 K and J∥ = 0.033 and 0.145 K. The proposed approach can be valuable in the absence of a more realistic mode even though the quantitative analysis of the exchange couplings should consider the Fe–X⋯Im⋯X–Fe exchange magnetic coupling. The estimated exchange parameters show that the Fe–X⋯X–Fe interactions along the b axis are the strongest. In addition, the least-squares fit of the experimental data yields weaker magnetic exchange coupling for the chloride compound, which confirms the macroscopic magnetic data.
 | ||
| Fig. 11 Temperature dependence of χm (black circles) for (a) Edimim[FeCl4] and (b) Edimim[FeBr4] measured under 1 kOe. The solid red lines are the fit according to eqn (2). | ||
Finally, it was supposed the bigger the imidazolium ion size on paramagnetic ionic liquids based on tetrahaloferrate ions, the weaker superexchange magnetic pathways, due to a larger distances between the iron–iron. However, comparison with Dimim[FeCl4]40 (5.6 K) and Dimim[FeBr4]41 (7.7 K) shows lower TN in the chloride compound (2.9 K) whereas it is higher for the bromide (9 K). On one hand, Fe⋯Fe distances in Dimim[FeBr4]41 are shorter; at 10 K they are 6.74 and 6.76 Å inside a layer and 8.57 Å between the layers. For Edimim[FeBr4] at 100 K, are 6.957 and 7.232 and 8.667 Å, respectively. On the other hand, the higher TN detected in Edimim[FeBr4] cannot be attributed to the geometrical factor of non-bonding interactions neither. Both bromide compounds, Dimim[FeBr4] and Edimim[FeBr4], show an almost linear coupling and out of-plane magnetic exchange angles in interplane and intraplane magnetic interactions, respectively with similar halide–halide distances. Thus, this enhancement of the TN with a bigger imidazolium cation in the bromide compounds will be attributed to the existence of a higher spin delocalization of iron atoms in the metal complex ions, [FeBr4]−, which favors the magnetic couplings.40,41 This results represents a new evidence of the relative importance of the spin density delocalization of the metal complex anion, which explains this higher efficiency in transmitting the magnetic interaction.
Conclusion
Two novel 3D magnetic correlated ILs based on imidazolium cation and tetrahaloferrate anion, namely Edimim[FeCl4] and Edimim[FeBr4], have been synthetized by solid-phase synthesis exhibiting melting points of 320 and 355 K for Cl and Br, respectively. The chloride compound, characterized by synchrotron X-ray powder diffraction at 100 K, crystallizes in the monoclinic space group P21/n. For the bromide composite, the crystal structure shows a monoclinic phase P21, characterized from single crystal X-ray diffraction at 100 K. Their frameworks, projected along b direction, are formed by layers of [Edimim]+ cations and [FeX4]− (X = Cl and Br) anions which change orientation from layer to layer, stacked upon one another in a 3D manner displaying three types of non-covalent interactions: hydrogen bonding, anion–π, and halide–halide. Raman measurements of both compounds at RT corroborate the presence of [FeX4]− (X = Cl and Br) and the gauche (non-planar) equilibrium conformation of the imidazolium cation with respect to NCC angle of the ethyl group. Magnetic studies of both compounds using magnetic susceptibility, magnetization and heat capacity measurements, show predominantly AF interactions and 3D magnetic ordering below 2.9 and 9 K for Edimim[FeCl4] and Edimim[FeBr4], respectively; together with the presence of stronger magnetocrystalline anisotropy in the latter. The magneto-structural correlations in both compounds give evidence that the 3D magnetic ordering mainly takes place via Fe–X⋯X–Fe interactions (X = Cl and Br), displaying a higher superexchange magnetic interaction between the planes. A comparative study with Dimim[FeX4] (X = Cl and Br) shows a lower TN in the chloride compound whereas it is higher for the bromide composite. This issue discards the hypothesis that the size of the cation play the most important role in the strength of the magnetic exchange pathways of the paramagnetic ionic liquids based on tetrahaloferrate anion and imidazolium cation.Acknowledgements
Financial support from the Spanish Ministerio de Ciencia e Innovación (Projects MAT2011-27573-C04) and Becas Iberoamericas Jóvenes Profesores Investigadores, 2015, Santander Universidades is acknowledged. The authors gratefully acknowledge the MALTA Consolider Ingenio 2010 (Ref. CSD2007-00045). IH acknowledges funding from the EU FP7 (Marie Curie-CIG 303535). X-Ray Synchrotron powder diffraction experiments were performed at BL04 beamline of the ALBA Synchrotron (proposal id 2013100582).References
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed.
- E. Santos, J. Albo and A. Irabien, RSC Adv., 2014, 4, 40008–40018 RSC.
- T. Torimoto, T. Tsuda, K.-i. Okazaki and S. Kuwabata, Adv. Mater., 2010, 22, 1196–1221 CrossRef CAS PubMed.
- M. Dobbelin, V. Jovanovski, I. Llarena, L. J. Claros Marfil, G. Cabanero, J. Rodriguez and D. Mecerreyes, Polym. Chem., 2011, 2, 1275–1278 RSC.
- B. Mallick, B. Balke, C. Felser and A.-V. Mudring, Angew. Chem., Int. Ed., 2008, 47, 7635–7638 CrossRef CAS PubMed.
- M. Okuhata, Y. Funasako, K. Takahashi and T. Mochida, Chem. Commun., 2013, 49, 7662–7664 RSC.
- A. Branco, L. C. Branco and F. Pina, Chem. Commun., 2011, 47, 2300–2302 RSC.
- A.-V. Mudring, A. Babai, S. Arenz, R. Giernoth, K. Binnemans, K. Driesen and P. Nockemann, J. Alloys Compd., 2006, 418, 204–208 CrossRef CAS PubMed.
- S. Hayashi and H.-o. Hamaguchi, Chem. Lett., 2004, 33, 1590–1591 CrossRef CAS.
- I. de Pedro, D. P. Rojas, J. Albo, P. Luis, A. Irabien, J. A. Blanco and J. Rodriguez Fernandez, J. Phys.: Condens. Matter, 2010, 22, 29006 CrossRef PubMed.
- I. de Pedro, D. P. Rojas, J. A. Blanco and J. Rodriguez Fernandez, J. Magn. Magn. Mater., 2011, 323, 1254–1257 CrossRef CAS PubMed.
- P. De Vreese, N. R. Brooks, K. Van Hecke, L. Van Meervelt, E. Matthijs, K. Binnemans and R. Van Deun, Inorg. Chem., 2012, 51, 4972–4981 CrossRef CAS PubMed.
- S. Pitula and A. V. Mudring, Chem.–Eur. J., 2010, 16, 3355–3365 CrossRef CAS PubMed.
- R. E. Del Sesto, T. M. McCleskey, A. K. Burrell, G. A. Baker, J. D. Thompson, B. L. Scott, J. S. Wilkes and P. Williams, Chem. Commun., 2008, 447–449 RSC.
- T. Peppel, M. Köckerling, M. Geppert-Rybczyńska, R. V. Ralys, J. K. Lehmann, S. P. Verevkin and A. Heintz, Angew. Chem., Int. Ed., 2010, 49, 7116–7119 CrossRef CAS PubMed.
- S. Tang, A. Babai and A.-V. Mudring, Angew. Chem., Int. Ed., 2008, 47, 7631–7634 CrossRef CAS PubMed.
- K. Binnemans, Chem. Rev., 2007, 107, 2592–2614 CrossRef CAS PubMed.
- K. Tanaka, F. Ishiguro and Y. Chujo, J. Am. Chem. Soc., 2010, 132, 17649–17651 CrossRef CAS PubMed.
- C.-X. Miao, J.-Q. Wang, B. Yu, W.-G. Cheng, J. Sun, S. Chanfreau, L.-N. He and S.-J. Zhang, Chem. Commun., 2011, 47, 2697–2699 RSC.
- O. Nacham, K. D. Clark, H. Yu and J. L. Anderson, Chem. Mater., 2015, 27, 923–931 CrossRef CAS.
- P. Brown, C. P. Butts, J. Eastoe, E. Padron Hernandez, F. L. d. A. Machado and R. J. de Oliveira, Chem. Commun., 2013, 49, 2765–2767 RSC.
- M. Okuno, H. Hamaguchi and S. Hayashi, Appl. Phys. Lett., 2006, 89, 132506 CrossRef PubMed.
- E. Santos, J. Albo, A. Rosatella, C. A. M. Afonso and Á. Irabien, J. Chem. Technol. Biotechnol., 2014, 89, 866–871 CrossRef CAS PubMed.
- P. Brown, A. Bushmelev, C. P. Butts, J. Cheng, J. Eastoe, I. Grillo, R. K. Heenan and A. M. Schmidt, Angew. Chem., Int. Ed., 2012, 51, 2414–2416 CrossRef CAS PubMed.
- K. D. Clark, O. Nacham, H. Yu, T. Li, M. M. Yamsek, D. R. Ronning and J. L. Anderson, Anal. Chem., 2015, 87, 1552–1599 CrossRef CAS PubMed.
- A. H. Mohammad Fauzi, N. A. S. Amin and R. Mat, Appl. Energy, 2014, 114, 809–818 CrossRef CAS PubMed.
- R. Giernoth, A. Bröhl, M. Brehm and Y. Lingscheid, J. Mol. Liq., 2014, 192, 55–58 CrossRef CAS PubMed.
- S. Zhang, J. Wang, X. Lu and Q. Zhou, Structures and Interactions of Ionic Liquids, Springer, 2014 Search PubMed.
- K. Fumino and R. Ludwig, J. Mol. Liq., 2014, 192, 94–102 CrossRef CAS PubMed.
- J. Dupont, Acc. Chem. Res., 2011, 44, 1223–1231 CrossRef CAS PubMed.
- R. P. Matthews, T. Welton and P. Hunt, Phys. Chem. Chem. Phys., 2015, 17, 14437–14453 RSC.
- J. Dupont, J. Braz. Chem. Soc., 2004, 15, 341–350 CrossRef CAS PubMed.
- S. Saha, S. Hayashi, A. Kobayashi and H.-o. Hamaguchi, Chem. Lett., 2003, 32, 740–741 CrossRef CAS.
- H. Weingärtner, Angew. Chem., Int. Ed., 2008, 47, 654–670 CrossRef PubMed.
- R. Bertani, P. Sgarbossa, A. Venzo, F. Lelj, M. Amati, G. Resnati, T. Pilati, P. Metrangolo and G. Terraneo, Coord. Chem. Rev., 2010, 254, 677–695 CrossRef CAS PubMed.
- A. Frontera, P. Gamez, M. Mascal, T. J. Mooibroek and J. Reedijk, Angew. Chem., Int. Ed., 2011, 50, 9564–9583 CrossRef CAS PubMed.
- Y. Yoshida, A. Otsuka, G. Saito, S. Natsume, E. Nishibori, M. Takata, M. Sakata, M. Takahashi and T. Yoko, Bull. Chem. Soc. Jpn., 2005, 78, 1921–1928 CrossRef CAS.
- Y. Yoshida and G. Saito, J. Mater. Chem., 2006, 16, 1254–1262 RSC.
- A. García-Saiz, I. de Pedro, J. A. Blanco, J. González and J. Rodríguez Fernández, J. Phys. Chem. B, 2013, 117, 3198–3206 CrossRef PubMed.
- A. García-Saiz, P. Migowski, O. Vallcorba, J. Junquera, J. A. Blanco, J. A. González, M. T. Fernández-Díaz, J. Rius, J. Dupont, J. Rodríguez Fernández and I. de Pedro, Chem.–Eur. J., 2014, 20, 72–76 CrossRef PubMed.
- A. Garcia-Saiz, I. de Pedro, P. Migowski, O. Vallcorba, J. Junquera, J. A. Blanco, O. Fabelo, D. Sheptyakov, J. C. Waerenborgh, M. T. Fernandez-Diaz, J. Rius, J. Dupont, J. A. Gonzalez and J. Rodríguez Fernández, Inorg. Chem., 2014, 53, 8384–8396 CrossRef CAS PubMed.
- S. Hayashi, S. Saha and H. Hamaguchi, IEEE Trans. Magn., 2006, 42, 12 CrossRef CAS.
- H. Tokuda, K. Hayamizu, K. Ishii, M. A. B. H. Susan and M. Watanabe, J. Phys. Chem. B, 2005, 109, 6103–6110 CrossRef CAS PubMed.
- A. Boultif and D. Louër, J. Appl. Crystallogr., 2004, 37, 724–731 CrossRef CAS.
- O. Vallcorba, J. Rius, C. Frontera, I. Peral and C. Miravitlles, J. Appl. Crystallogr., 2012, 45, 844–848 CrossRef CAS.
- O. Vallcorba, J. Rius, C. Frontera and C. Miravitlles, J. Appl. Crystallogr., 2012, 45, 1270–1277 CrossRef CAS.
- I. J. Bruno, J. C. Cole, M. Kessler, J. Luo, W. S. Motherwell, L. H. Purkis, B. R. Smith, R. Taylor, R. I. Cooper and S. E. Harris, J. Chem. Inf. Comput. Sci., 2004, 44, 2133–2144 CrossRef CAS PubMed.
- Bruker, SADABS, Bruker AXS Inc, Madison, Wisconsin, USA, 2001 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- D. Appleby, C. L. Hussey, K. R. Seddon and J. E. Turp, Nature, 1986, 323, 614–616 CrossRef CAS PubMed.
- Y. Yoshida and G. Saito, J. Mater. Chem., 2006, 16, 1254–1262 RSC.
- C. Zhong, T. Sasaki, A. Jimbo-Kobayashi, E. Fujiwara, A. Kobayashi, M. Tada and Y. Iwasawa, Bull. Chem. Soc. Jpn., 2007, 80, 2365–2374 CrossRef CAS.
- Y. Umebayashi, T. Fujimori, T. Sukizaki, M. Asada, K. Fujii, R. Kanzaki and S.-i. Ishiguro, J. Phys. Chem. A, 2005, 109, 8976–8982 CrossRef CAS PubMed.
- J. Estager, J. Holbrey and M. Swadźba-Kwaśny, Chem. Soc. Rev., 2014, 43, 847–886 RSC.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- B. P. Hay and R. Custelcean, Cryst. Growth Des., 2009, 9, 2539–2545 CAS.
- J.-Y. Kim, J.-T. Kim, E.-A. Song, Y.-K. Min and H.-o. Hamaguchi, Macromolecules, 2008, 41, 2886–2889 CrossRef CAS.
- P. Larkin, Infrared and Raman spectroscopy; principles and spectral interpretation, Elsevier, 2011 Search PubMed.
- R. Ozawa, S. Hayashi, S. Saha, A. Kobayashi and H.-o. Hamaguchi, Chem. Lett., 2003, 32, 948–949 CrossRef CAS.
- N. E. Heimer, R. E. Del Sesto, Z. Meng, J. S. Wilkes and W. R. Carper, J. Mol. Liq., 2006, 124, 84–95 CrossRef CAS PubMed.
- I. de Pedro, A. Garcia-Saiz, J. Gonzalez, I. Ruiz de Larramendi, T. Rojo, C. A. M. Afonso, S. P. Simeonov, J. C. Waerenborgh, J. A. Blanco, B. Ramajo and J. Rodríguez Fernández, Phys. Chem. Chem. Phys., 2013, 15, 12724–12733 RSC.
- I. de Pedro, J. M. Rojo, J. Rodríguez Fernández, M. T. Fernández-Díaz and T. Rojo, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 134431 CrossRef.
- I. de Pedro, J. M. Rojo, J. Rodríguez Fernández, J. S. Marcos, M. T. Fernandez-Diaz and T. Rojo, J. Solid State Chem., 2012, 188, 1–10 CrossRef CAS PubMed.
- P. Debye, Ann. Phys., 1912, 344, 789–839 CrossRef PubMed.
- I. De Pedro, J. M. Rojo, J. Rius, O. Vallcorba, I. R. De Larramendi, J. Rodríguez Fernández, L. Lezama and T. Rojo, Inorg. Chem., 2012, 51, 5246–5256 CrossRef CAS PubMed.
- I. de Pedro, J. M. Rojo, J. Rodríguez Fernández, L. Lezama and T. Rojo, Eur. J. Inorg. Chem., 2010, 17, 2514–2522 CrossRef PubMed.
- J. Glerup, P. A. Goodson, D. J. Hodgson and K. Michelsen, Inorg. Chem., 1995, 34, 6255–6264 CrossRef CAS.
- J. Campo, J. Luzón, F. Palacio, G. J. McIntyre, A. Millán and A. R. Wildes, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 054415 CrossRef.
- J. McElearney, S. Merchant and R. Carlin, Inorg. Chem., 1973, 12, 906–908 CrossRef CAS.
- G. R. Wagner and S. A. Friedberg, Phys. Lett., 1964, 9, 11–13 CrossRef CAS.
- J. C. Bonner and M. E. Fisher, Phys. Rev., 1964, 135, A640–A658 CrossRef.
- G. S. Rushbrooke and P. J. Wood, Mol. Phys., 1958, 1, 257–283 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic information, CIF data, and supplementary magnetic measurements. CCDC 1056065. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra05723j |
| This journal is © The Royal Society of Chemistry 2015 |