
Acetaldehyde in asymmetric organocatalytic transformations
Abstract
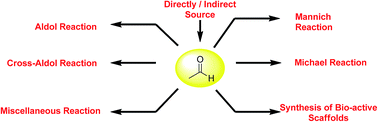
The role of acetaldehyde as a nucleophile in various asymmetric C–C bond forming transformations is presented, with consideration given not only to the optimization of reaction parameters relevant to product formation, polymerization, by-products, purification, chirality and instability of the final products, but also to the application of the final products in the synthesis of bioactive molecules. This review is organized according to the use of acetaldehyde in different organocatalytic reactions covering the most recent reports. In the last section indirect sources of acetaldehyde are discussed from the perspective of difficult handling and its requirement of slow addition as well as being freshly distilled in some of the transformations, which is one of the most critical issues in the late exploitation of acetaldehyde as a nucleophile in synthetic chemistry.
 Please wait while we load your content...
Please wait while we load your content...

