DOI:
10.1039/C5RA05651A
(Paper)
RSC Adv., 2015,
5, 37780-37788
Synthesis of polyacrylonitrile by reversible-deactivation radical polymerization and its application as electrode materials for electrochemical double layer capacitors
Received
31st March 2015
, Accepted 20th April 2015
First published on 20th April 2015
Abstract
Bis(acetylacetonato) iron(II) (Fe(acac)2) was first selected to mediate the reversible-deactivation radical polymerization (RDRP) of acrylonitrile (AN) with predictable molecular weights, relatively narrow molecular weight distributions and high end-fidelity. The reversible termination (RT) mechanism accounted well for the reaction phenomena. The spinning performance of polyacrylonitrile (PAN) was investigated and the average diameter of the spinning fibers was approximately 300 nm. Activated carbon fibers (ACFs) were prepared via sodium hydroxide (NaOH) direct activation. The maximum specific surface area of the ACF was as high as 1165 m2 g−1. The pore volume of the ACF reached 0.14 cm3 g−1. Most is the contribution of the small mesopores (2–5 nm). The unique microstructures enabled the ACFs to have good compatibility with potassium hydroxide (KOH) as electrolyte. The highest capacitance reached 167 F g−1 at room temperature. The electrode showed well stabilized capacitance after 500 cycles and high energy/power density values due to the formation of small mesopores.
Introduction
Electrochemical double layer capacitors (EDLCs) are the preferred alternative energy sources to solve the energy crisis due to their high power density and long cycle life.1–3 For example, they have played vital roles in the field of high-performance electronic products.4,5 Whereas, as the bottlenecks, low energy density and high cost have restricted the development of EDLCs.6,7 Hence development of novel electrode materials is an efficient approach to solve these issues. Carbon materials have attracted much attention for their high specific surface areas. Currently, they can be classified into activated carbon,8 carbon aerogel,9 carbon nanotubes,10 and activated carbon fibers (ACFs).11 Polyacrylonitrile (PAN)-based ACFs have distinguished themselves for chemical stability, environmental compatibility, excellent electroconductivity, and high porosity. Theoretically, the higher is specific surface area of the carbon, the better will be its competence for charges collection.12,13 Therefore, as the EDLCs electrode materials, PAN-based ACFs can provide well reaction paths.
To date, all the precursors of PAN-based ACFs are prepared by traditional free radical polymerization (RP) techniques. These methods generally tend to cause broken filament of PAN during the electrospinning process. It may exert a negative impact on the performance of EDLCs. On the other hand, the time for pre-oxidation procedure will be shortened if the diameter of PAN nanofibers is relatively slenderer. Meanwhile, fine-denier filament is one of principal pathways to obtain high-performance PAN-based ACFs.14 As a result, it is of vital importance to employ a proper polymerization technique to prepare the precursor of PAN-based ACFs with appropriate molecular weights (Mw) and relatively narrower molecular weight distributions (MWDs). Compared with the RP methods, reversible-deactivation radical polymerization (RDRP) techniques15–18 can decrease lousiness, tangle and fracture of nanofibres via electrospinning due to inherent excellent control over Mw and MWDs of polymers.
Note that transition metal catalysts have been widely used in RDRP methods, especially iron,17 its halogen compounds18 and organic compounds19 in light of their low cost and biocompatibility. Iron(0) has been successfully used to polymerize acrylonitrile (AN) as the catalyst based on single electron transfer-living radical polymerization (SET-LRP) in which MWDs maintained in the range of 1.3–1.5.20 It is also demonstrated to mediate polymerization of methyl methacrylate (MMA) in combination with 2-cyanoprop-2-yl 1-dithionaphthalate (CPDN), which is one typical reversible addition-fragmentation chain transfer (RAFT) agent.21,22 Intermediate-valence iron/ligand has catalyzed MMA polymerization with an atom transfer radical polymerization (ATRP) initiator, rendering relatively narrower MWDs of 1.1–1.3.23,24 The highest oxidation state iron(III), has been found as an active catalyst in activators regenerated by electron transfer ATRP (ARGET ATRP)25 with less usage than that in activators generated by electron transfer ATRP (AGET ATRP).26
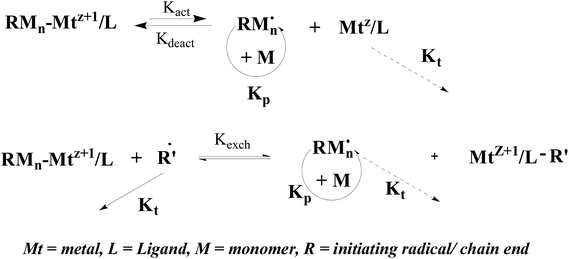
However, the RDRP technique is still rather limited in terms of choice of iron, with only a handful of its complexes having proven successful. Actually, organometallic compounds have also shown great charm since cobalt tetramesitylporphyrin (TMP-Co) perfectly mediated methyl acrylate (MA) polymerization reported by Wayland.27 Generally, polymerization of vinyl monomers mediated by organometallic compound mainly based on two mechanisms provided that they contain sufficiently weak, and thermally fragile metal–carbon bonds, as shown in Scheme 1.28 Bis(acetylacetonato) iron(II) (Fe(acac)2), as one typical organometallic compound, has shown good coordination for its distinct molecular structure.29 Greatly different from the catalyst role previously acted by iron(0) and its halogen compounds, Fe(acac)2 was firstly used as a reversible radical capping agent to polymerize VAc with narrow MWDs (1.29–1.36) in Poli's research group.30
 |
| | Scheme 1 Mechanisms of organometallic mediated radical polymerization. | |
In consideration of broadening Fe(acac)2 field in RDRP techniques, PAN was firstly synthesized in the presence of Fe(acac)2. Application of the commercial available Fe(acac)2 possesses potential values for its environmental compatibility and nontoxicity. PAN nanofibers were obtained via electrospinning using the PAN/N,N-dimethylformamide (DMF) homogeneous solution. To simplify the subsequent procedures and save cost, PAN-based ACFs were yielded by sodium hydroxide (NaOH) direct activation. The macrostructures of PAN-based ACFs were characterized in detail. The evaluation of electrochemical performance of the PAN-based ACFs was investigated with potassium hydroxide (KOH) as electrolyte, avoiding drawbacks of aprotic electrolyte.31–33
Result and discussion
“Living”/controlled radical polymerization of AN with Fe(acac)2 as the mediator
Until now only one report notes that bis(acetylacetonate) cobalt(II) (Co(acac)2) has been successfully mediated the “living”/controlled radical polymerization of AN concerning to the aspect of organic metal salts.34 To broaden their species in polymerizing AN, it is interesting to test the commercial available and environmental compatible Fe(acac)2 for the radical polymerization of AN. The polymerization results are summarized in Table 1. It was found that the polymerization did not take place even after 2 days without AIBN (entries 1 and 2 in Table 1). While the monomer conversion reached 27.70% after 2 h with only a handful of AIBN (entry 3 in Table 1). However, the polymerization showed typical radical polymerization behavior without Fe(acac)2 for the uncontrolled molecular weights (Mn = 149![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700) and a wide molecular weight distribution (Mw/Mn = 2.60). When the polymerization was conducted with both AIBN and Fe(acac)2, the obtained polymers showed controlled molecular weights and relatively narrower molecular weight distributions, which are shown in entries 4–12 in Table 1. These initial investigations show that the polymerization of AN can be controlled to some extent with AIBN as the initiator and Fe(acac)2 as mediator. To further investigate the polymerization behavior, the polymerization kinetics of AN with different molar ratios of AIBN was shown in Fig. 1. The results demonstrate that the monomer consumption lived up to the first order rate law. Based on the equation of the semilogarithmic plot ln([M]0/[M]) as a function of time (ln([M]0/[M]) = kp[R˙]t), a conclusion can be drawn that the concentration of active species was almost constant, and the probable occurrence of the irreversible transfer reactions or termination was almost equal to zero. An induction time was observed since no obvious resulting polymer appeared, indicating the occurrence of radical trapping by Fe(acac)2. The induction time can be reduced by increasing the amount of initiator, which was confirmed in the investigation in entries 4 and 5, 7 and 8, 10 and 11 in Table 1, e.g., ∼1 h, ∼0.5 h and ∼0.3 h in the cases of [AN]0:[Fe(acac)2]0:[AIBN]0 = 200:1:0.5, 200:1:1 and 200:1:1.5, respectively. The derivation of molecular weights and molecular weights distributions with AN conversion is shown in Fig. 2. The molecular weights of PAN increased with conversion and the range molecular weight distributions was 1.40–1.59. All of this evidence indicates that the polymerization of AN was well-controlled using AIBN as the initiator and Fe(acac)2 as the mediator.
700) and a wide molecular weight distribution (Mw/Mn = 2.60). When the polymerization was conducted with both AIBN and Fe(acac)2, the obtained polymers showed controlled molecular weights and relatively narrower molecular weight distributions, which are shown in entries 4–12 in Table 1. These initial investigations show that the polymerization of AN can be controlled to some extent with AIBN as the initiator and Fe(acac)2 as mediator. To further investigate the polymerization behavior, the polymerization kinetics of AN with different molar ratios of AIBN was shown in Fig. 1. The results demonstrate that the monomer consumption lived up to the first order rate law. Based on the equation of the semilogarithmic plot ln([M]0/[M]) as a function of time (ln([M]0/[M]) = kp[R˙]t), a conclusion can be drawn that the concentration of active species was almost constant, and the probable occurrence of the irreversible transfer reactions or termination was almost equal to zero. An induction time was observed since no obvious resulting polymer appeared, indicating the occurrence of radical trapping by Fe(acac)2. The induction time can be reduced by increasing the amount of initiator, which was confirmed in the investigation in entries 4 and 5, 7 and 8, 10 and 11 in Table 1, e.g., ∼1 h, ∼0.5 h and ∼0.3 h in the cases of [AN]0:[Fe(acac)2]0:[AIBN]0 = 200:1:0.5, 200:1:1 and 200:1:1.5, respectively. The derivation of molecular weights and molecular weights distributions with AN conversion is shown in Fig. 2. The molecular weights of PAN increased with conversion and the range molecular weight distributions was 1.40–1.59. All of this evidence indicates that the polymerization of AN was well-controlled using AIBN as the initiator and Fe(acac)2 as the mediator.
Table 1 The polymerization of AN with different amounts of AIBN mediated by Fe(acac)2 at 60 °C
| Entry |
[AN]0:[Fe(acac)2]0:[AIBN]0 |
Time (h) |
Conv.% |
Mn (g mol−1) |
Mw/Mn |
| 1 |
200:1:0 |
24 |
0 |
— |
— |
| 2 |
48 |
0 |
— |
— |
| 3 |
200:0:1 |
2 |
27.70 |
149700 |
2.60 |
| 4 |
200:1:0.5 |
0.5 |
0 |
— |
— |
| 5 |
1 |
0.55 |
104800 |
1.46 |
| 6 |
2 |
15.21 |
155200 |
1.54 |
| 7 |
200:1:1.0 |
0.3 |
0 |
— |
— |
| 8 |
0.5 |
1.07 |
95400 |
1.44 |
| 9 |
1 |
15.03 |
115300 |
1.51 |
| 10 |
200:1:1.5 |
0.17 |
0 |
— |
— |
| 11 |
0.3 |
0.41 |
75200 |
1.48 |
| 12 |
0.5 |
2.85 |
84100 |
1.45 |
 |
| | Fig. 1 First order kinetics plots of polymerization of AN mediated by Fe(acac)2 at 60 °C under the conditions of [AN]0:[Fe(acac)2]0:[ AIBN]0 = 200:1:■ 0.5; ● 1.0; ▲ 1.5, and AN:DMF = 1:1 (v/v). All the conditions were executed with 1 mL of AN. [M]0 and [M] referred to the initial concentration and instantaneous concentration of AN, respectively. | |
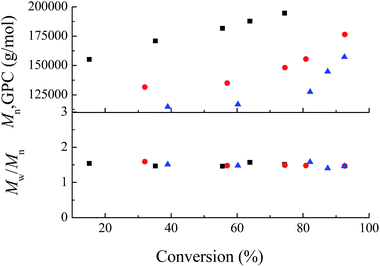 |
| | Fig. 2 The number-average molecular weights (Mn) and molecular weight distributions (Mw/Mn) of resulting PAN initiated by various concentrations of AIBN in the presence of Fe(acac)2 at 60 °C (■ [AN]0: [Fe(acac)2]0:[AIBN] = 200:1:0.5, ● [AN]0:[Fe(acac)2]0:[AIBN] = 200:1:1.0, ▲ [AN]0:[Fe(acac)2]0:[AIBN] = 200:1:1.5). | |
As was reported in Poli's research group in terms of polymerization of VAc using Fe(acac)2, reversible termination (RT) occurred when addition of Lewis base such as DMF and pyridine that can act as ligands and in the absence of an excess amount of radicals.29 It was also consistent with the phenomenon that was investigated in his earlier research on polymerization of AN mediated by Co(acac)2.34 According to the RT mechanism (Scheme 1(a)), the molecular weights of PAN can be tuned both by changing molar ratio between AN and Fe(acac)2 and by monomer conversion. In order to verify such a mechanism in the current polymerization system, three experiments were run with different ratios of [AN]0/[Fe(acac)2]0, keeping [Fe(acac)2]0/[AIBN]0 = 1:1 at 60 °C. The results are summarized in Fig. 3 and 4. From the kinetics point of view, the three experiments presented the same behavior, i.e., the time dependence of the ln([M]0/[M]) function was linear. The polymerization rate was visibly decreased due to the lower amounts of AIBN. On the other hand, the monomer conversion was increased linearly versus time. Concerning to RT mechanism, the PAN molecular weights will strongly depend on the [AN]0/[Fe(acac)2]0 for a given monomer conversion. Fig. 4 shows that contrast of Mn in all the cases were consistent with the RT mechanism, i.e., Mn ([AN]0/[Fe(acac)2]0 = 400:1) > Mn ([AN]0/[Fe(acac)2]0 = 200:1) > Mn ([AN]0/[Fe(acac)2]0 = 100:1) once the AN conversion was the same. Note that also in this case the polymerization was sustained well beyond the time needed to generate new radicals, confirming the reversible release of growing chains from the dormant species according to RT mechanism. The linear increased Mn with conversion and relatively narrow Mw/Mn (Mw/Mn < 1.60) further manifested the controllable characteristic of polymerization mediated by Fe(acac)2.
 |
| | Fig. 3 Time dependence of ln([M]0/[M]) and conversion for the AN polymerization initiated by AIBN in DMF at 60 °C using different [AN]/[Co(acac)2] ratios (■ [AN]0/[Fe(acac)2]0 = 100:1, ● [AN]0/[Fe(acac)2]0 = 200:1, ▲ [AN]0/[Fe(acac)2]0 = 400:1). All the experiments were conducted with AN = 1 mL and AN:DMF = 1:1 (v/v). | |
 |
| | Fig. 4 The plots of conversion versus the number-average molecular weights (Mn) and molecular weight distributions (Mw/Mn) of resulting PAN obtained from Fe(acac)2-mediated polymerization at 60 °C (■ [AN]0:[Fe(acac)2]0:[AIBN] = 100:1:1, ● [AN]0:[Fe(acac)2]0:[AIBN] = 200:1:1, ▲ [AN]0:[Fe(acac)2]0:[AIBN] = 400:1:1). | |
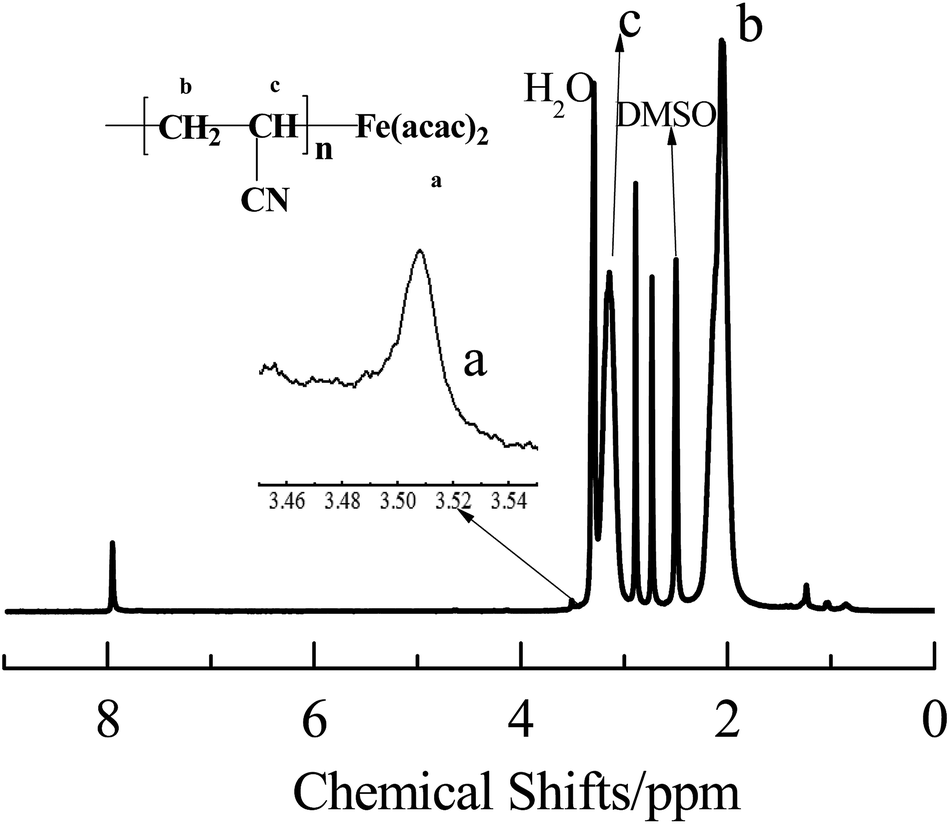
The chain end-fidelity of PAN sample prepared in the presence of AIBN and Fe(acac)2 was analyzed by 1H NMR spectrum (Fig. 5). The PAN sample was obtained at 32.0% conversion from the polymerization with 200:1:1 of [AN]0:[Fe(acac)2]0:[AIBN]0 and 1:1 of [AN]:[DMF] (v/v). The signals at δ = 2.15 ppm, and 3.24 ppm were attributed to –CH2 and –CH (CN) in the main chains of PAN, respectively. The signals at δ = 3.48–3.52 ppm was corresponded to protons of acetyl acetone groups, indicating that Fe(acac)2 was successfully end-capped on the PAN chain. The chemical shifts demonstrate that polymerization was proceeded in a controlled manner. Based on the above discussion, the mechanism of polymerization of AN with AIBN as the initiator and Fe(acac)2 as the mediator can be proposed, as shown in Scheme 2.
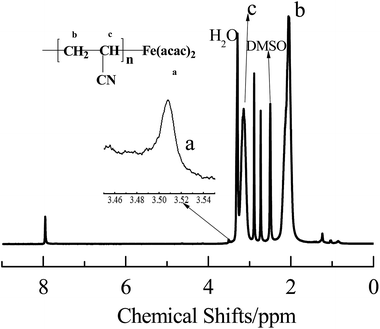 |
| | Fig. 5 1H NMR spectra of the resulting PAN of 12 h mediated by Fe(acca)2 under the conditions of [AN]0:[Fe(acac)2]0:[AIBN] = 200:1:1, and AN:DMF = 1:1 (v/v) at 60 °C, with DMSO-d6 as the solvent. | |
 |
| | Scheme 2 Polymerization mechanism for AN using AIBN as the initiator and Fe(acac)2 as the mediator in DMF. | |
Preparation and characterization of ACFs
The spinnability of the pre-prepared PAN homopolymers, which were obtained under the conditions of [AN]0:[Fe(acac)2]0:[AIBN]0 = 200:1:1, and AN/DMF = 1:1 (v/v) for 12 h, was investigated via electrospinning setup. The images of the nanofiber were presented by the SEM, as assessed by Fig. 6(A) and (a). All the samples manifested cylindrical morphologies, and were long enough and continuous without breakage. The diameter of nanofibers was mostly distributed at 300 nm, much finer and more homogeneous compared with those reported in ref. 4. Fig. 6(B) and (b) shows the trapezoidal frame image of PAN after the pre-oxidation period. It is corresponding to the reactions of cyclization, dehydrogenation and oxidation. The diameter and surface of the pre-oxidized fiber in Fig. 6(b), compared with those in Fig. 6(a), became relatively larger and rougher, respectively, for pyrolysis of a fraction of nanofiber. Fig. 6(C) and (c) exhibits the surface morphology of ACF-1. It can be obviously seen that the char was etched off. With increasing NaOH content for activation, as assessed by Fig. 6(D, d) and (E, e), more fibers of ACF-2 and ACF-3 were tangled and etching fragments of large numbers of fibers became obvious.
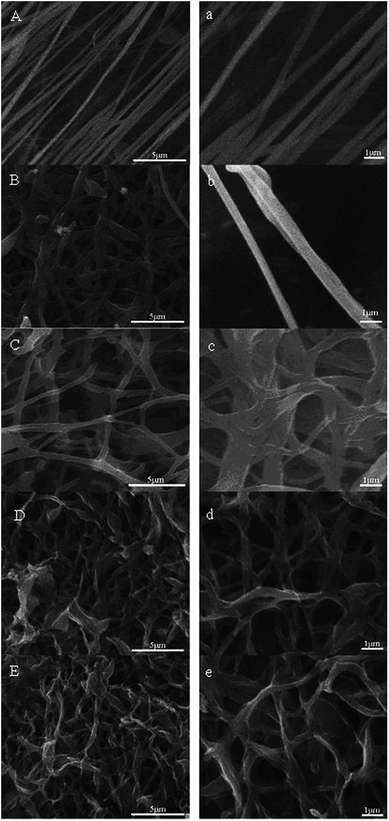 |
| | Fig. 6 SEM images of nanofibers fabricated by electrospinning (A and a); nanofibers obtained from preoxidation (B and b); ACF-1 (C and c); ACF-2 (D and d); ACF-3 (E and e). | |
Fig. 7 exhibits XRD spectrograms of PAN powder (a), pre-oxidized nanofiber (b), ACF-1 (c), ACF-2 (d), and ACF-3 (e). The two X-ray diffraction peaks, 2θ = 16.9° which was intensive and 2θ = 28.9° which was relatively weaker in Fig. 7(a), were conformed to those of PAN powder in publications.35 A new X-ray diffraction peak appeared approximately at 2θ = 25° in Fig. 7 (b–e), compared with Fig. 7(a), indicating the formation and stability of the polyimide trapezoidal frame after pre-oxidation process and carbonation process. The intensities of both wide X-ray diffraction peaks, approximately at 2θ = 25° and 45°, became gradually weak varied from (c) to (e) in Fig. 7, indicating the crystallization and orientation fading away. It provides a further evidence for the formation of amorphous carbon materials.
 |
| | Fig. 7 The XRD patterns of PAN powder (a); nanofibers obtained from preoxidation (b); ACF-1 (c); ACF-2 (d); ACF-3 (e). | |
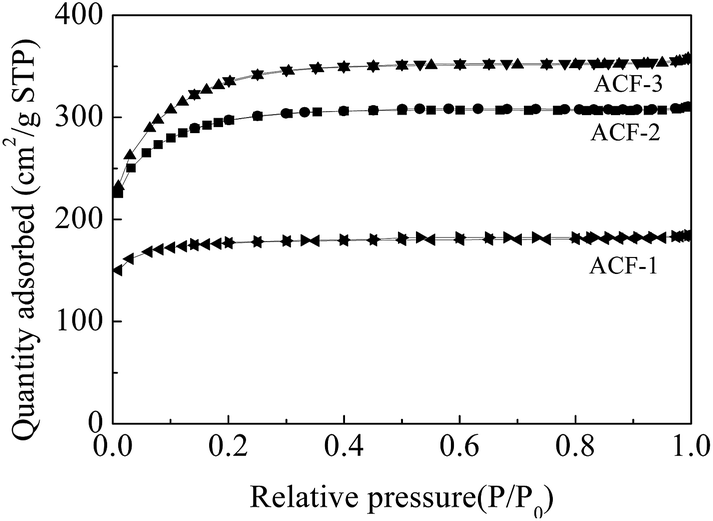
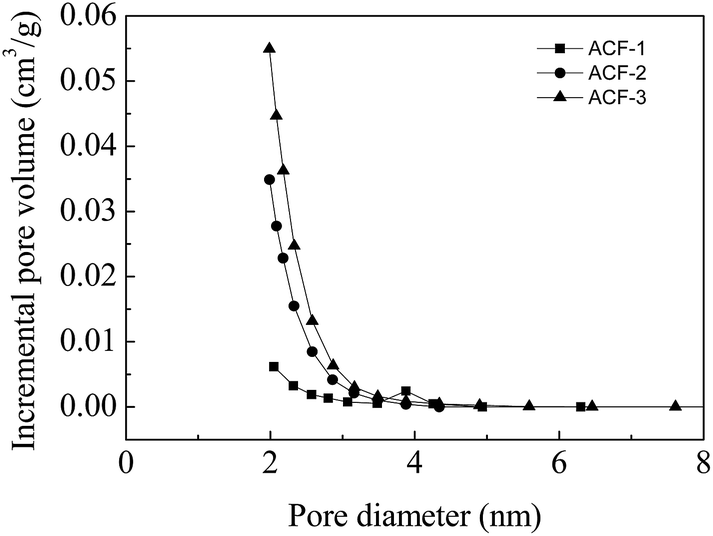
Generally, chemical activation was composed of two processes, i.e., pore formation and pore widening. Fig. 8 shows the N2 adsorption/desorption isotherms of the ACFs at 77 K. All the isotherms of ACF-1, ACF-2 and ACF-3 were type I according to the IUPAC classification. The pore volumes were mostly filled below P/P0 ≈ 0.1, indicating that micropores played a predominant role. The nitrogen adsorption capacity and BET surface area increased (595–1165 m2 g−1) as the activation rates increased. Though it is a preliminary attempt that the precursor PAN of ACFs was synthesized by Fe(acac)2-mediated RDRP technique, the specific surface areas were comparable with those reported by Kap Seung Yang.36 According to the pore size distributions (Fig. 9), the mesopore volume fraction increased from ACF-1 to ACF-3, indicating that mesopores (2–5 nm) formed through the unification of micropores at elevated activation ratios. The maximum mesopore volume was 0.14 cm3 g−1 of ACF-3, providing a well path for electrons movement.
 |
| | Fig. 8 N2 adsorption/desorption isotherms of the ACF samples according to BET method. | |
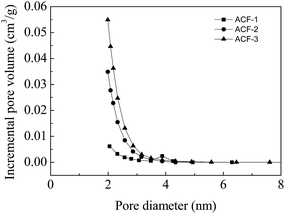 |
| | Fig. 9 Pore size distributions according to DFT method. | |
Electrochemical performance of ACFs
The porous ACFs, i.e., ACF-1, ACF-2, and ACF-3, were attached to square nickel foam to evaluate the capacitances in a three-electrode system, respectively. Here, 6 M KOH solution was selected to be the electrolyte. Fig. 10 shows the typical cyclic voltammetry (CV) curves at the similar potential ranges. The phenomena that the three CV curves presented quasi rectangle without any obvious redox peak, as shown in Fig. 10, were attributed to formation of surface functional groups owning to NaOH activation. That is a distinguished feature of EDLCs as well. The capacitances of ACF-1, ACF-2, and ACF-3 were 77 F g−1, 101 F g−1, and 123 F g−1, respectively. It is amply demonstrated that these mesopores of ACF-3 played dominant role for providing available access for KOH. Therefore, ACF-3 was the optimum candidate of the EDLC in the subsequent test. Fig. 11 exhibited CV curves at various scan rates in the same potential range (−1.4 V to 0.5 V). The CV curves revealed as quasi-rectangular without any obvious redox peaks. The maximum capacitance was 167 F g−1 when the scan rate was 30 mV s−1, much higher than that (111 F g−1) of carbon nanotubes grafted PAN-based activated carbon cloth as the electrode.37 All the discussion above demonstrate that the EDLCs exhibited excellent capacitive performance with the PAN precursor synthesized by Fe(acac)2-mediated RDRP method. Impedance of the ACF-1, ACF-2, and ACF-3 were measured in the frequency range of 100 kHz to 1 Hz at the ambient temperature, as shown in Fig. 12. It could be obviously seen that the impedance spectra are almost similar in form, composed of one semicircle at high-frequency which equals to the charge transfer resistance and a linear part at the low-frequency. However, the semicircle diameter of ACF-3 was smaller than those of ACF-1 and ACF-2. The slope of the linear part of ACF-3 was larger than those of ACF-1 and ACF-2. As a result, a conclusion can be drawn that ACF-3 was more closer to the ideal EDLC. The long-term cycle stability of ACF-3 was evaluated in this study during the charge/discharge process with a voltage of 1.6 V at a current density of 6 A g−1 in 6 M KOH electrolyte (Fig. 13). ACF-3 exhibited relatively better electrochemical stability after 500 cycles. It can be seen from Fig. 13 that the linear potential-time dependence demonstrated the typical capacitive behavior of the electrode. The redox reactions can be ignored. The calculated capacitance as a function of the current density was shown in Fig. 14. It can be seen that the rate capability of ACF-2 and ACF-3 were better than ACF-1. The capacitance retention were 57.9%, 73.52% and 67.0% of ACF-1, ACF-2 and ACF-3, respectively, relatively higher than those of Zn-substitution α-Co(OH)2 nanosheet electrode for EDLCs.38 As is known to all that power density and energy density are important parameters for the investigation of electrochemical performance of EDLCs. Fig. 15 exhibits the Ragone plot of ACF-based EDLCs measured at the ambient temperature. Compared with those of ACF-1 and ACF-2, the energy density of ACF-3 decreased slowly with an increase in power density. It can be obviously seen from Fig. 15 that efficient energy storage devices were produced using the ACF-3 electrode.
 |
| | Fig. 10 Typical cyclic voltammetry (CV) curves of the ACF-based EDLCs using KOH as the electrolyte (ACF-1: voltage: −1.3 V to 0.5 V, scan rate: 20 mV s−1, weight: 0.03521 g; ACF-2: voltage: −1.2 V to 0.4 V, scan rate: 10 mV s−1, weight: 0.02961 g; ACF-3: voltage: −1.2 V to 0.4 V, scan rate: 10 mV s−1, weight: 0.06293 g). | |
 |
| | Fig. 11 Typical CV curves of ACF-based EDLCs (ACF-3 was used here) using KOH as the electrolyte at different scan rates (voltage: −1.4 V to 0.5 V). | |
 |
| | Fig. 12 Nyquist plots of ACF-based EDLCs in the frequency range of 100 kHz to 1 Hz measured at the ambient temperature. | |
 |
| | Fig. 13 Retention of the specific capacitance of ACF-3 as a function of cycle number during the charge/discharge process with a voltage of 1.6 V at a current density of 6 A g−1 in 6 M KOH electrolyte. | |
 |
| | Fig. 14 Plots of capacitance as a function of discharge current density. | |
 |
| | Fig. 15 Ragone plot of ACF-based EDLCs in 6 M KOH electrolyte at ambient temperature. | |
Experimental
Materials
Acrylonitrile (AN) (>98.5%, Tianjin Fuchen Chemical Reagent Co., Tianjin, China) was purified via atmospheric distillation prior to being conserved at 5 °C. 2,2′-Azodiisobutyronitrile (AIBN) (A. R. grade, Tianjin Fuchen Chemical Reagent Co., Tianjin, China) was purified after recrystallization. Bis(acetylacetonato) iron(II) (Fe(acac)2) (>98.0%, Aladdin) was used without further purification. N,N-Dimethylformamide (DMF) (>99.5%, Tianjin Bodi Chemical Reagents Co., Tianjin, China) was dealt with molecular sieves to eliminate water. Nickel foam (>99.8%, Taiyuan yingze Lizhiyuan battery sales department, Taiyuan, China) was used after removal of oxide by ultrasonic treatment. Methanol (A. R. grade, Tianjin Damao Chemical Reagent Co., Tianjin, China), lithium bromide (LiBr) (>98.0%, Tianjin Guangfu Fine Chemical Institute, Tianjin, China), polyvinylidene fluoride (PVDF) (>99.8%, Taiyuan yingze Lizhiyuan battery sales department, Taiyuan, China), N-methyl pyrrolidone (>99.0%, Tianjin Damao Chemical Co., Tianjin, China), potassium hydroxide (KOH) (A. R. grade, Tianjin Ruijinte Chemical Reagents Co., Tianjin, China), sodium hydroxide (NaOH) (A. R. grade, Laiyang Economic and Technological Development Zone Fine Chemical Co., Yantai, China), nitrogen (N2) (99.999%, Yantai Feiyuan Gas Co., Yaitai, China), and hydrochloric acid (HCl) (A. R. grade, Laiyang Economic and Technological Development Zone Fine Chemical Co., Yantai, China) were used as received.
Apparatus and instrumentation
The number-average molecular weights (Mn) and molecular weight distributions (Mw/Mn) of obtained polymers were determined by Waters 1515 gel permeation chromatograph (GPC) assembled with a refractive-index detector (Waters 2414) using HR column (7.8 × 300 mm). DMF with 0.1 M LiBr was used as the eluent at a flow rate of 1.0 mL min−1 and 50 °C. The 1H NMR spectrum of the precipitated polymers was recorded on an INOVA 400 MHz nuclear magnetic resonance instrument using DMSO-d6 as the solvent. The non-woven mats were obtained by the electrospinning setup (Beijing Fuyouma Science and Technology Ltd., Beijing, China), mainly composed by a 10 mL glass syringe, needle tips with different sizes, a high voltage power supply, and a stainless steel drum as the collector. The surface morphologies of samples were characterized by high and low vacuum scanning electron microscopy (SEM, JSM-5610LV, Tokyo, Japan), operating at 20 kV. The fiber of electrostatic spinning was sputter-coated with Au film prior to measurements. X-ray diffraction (XRD, D/max-2500PC, Japan) was used to analyze the crystal structure of samples. The specific surface areas were calculated based on BET method, and ASAP 2020N Physisorption Analyzer (American, Micromeritics) was employed. The electrochemical measurements were carried out using CHI 660C electrochemical workstation (Shanghai Chenhua Instrument Co., Shanghai, China).
Typical procedures for polymerization of AN in the presence of AIBN and Fe(acac)2
The monomer (AN, 1.0 mL, 15.19 mmol), initiator (AIBN, 12.5 mg, 0.076 mmol), end-capped agent (Fe(acac)2, 19.4 mg, 0.077 mmol) and solvent (DMF, 1.0 mL) were added to a 5.0 mL ampule in the following order: AIBN, Fe(acac)2, AN, and DMF. The solution was deoxygenated via three freeze–pump–thaw cycles. The ampule was flame-sealed subsequently and placed in a DF-101S magnetic stirred oil bath equipped with a thermostat at 60 ± 0.2 °C. The ampule was taken out from the oil bath and put into the ice water when it reached setting time. Then it was opened and the content was dissolved in 5–8 mL DMF. The resulting solution was precipitated in approximately 150 mL methanol with stirring. The polymer was separated via filtration and put in vacuum drying oven to remove the unreacted monomer at 60 °C until constant weight was recorded at room temperature. The monomer conversion was determined gravimetrically and the polymer was used for GPC and 1H NMR characterization.
The procedures of electrospinning and preparation of the ACFs
The nanofiber based on PAN homopolymer was prepared by a simple electrospinning process. The concentration of PAN in DMF was set to be 8%. A voltage of 18 kV and a tip-to-collector of 15 cm were applied to produce a web. The nanofiber was heated in the air at 200 °C for 2 h with a heating rate of 1 °C min−1. The pre-oxidized nanofiber was thoroughly soaked in a certain concentration of NaOH for several hours. It was dried at 60 °C in vacuum for 12 h. The fiber soaked in NaOH was directly activated at 600 °C for 1.5 h with a heating rate of 5 °C min−1 in tube furnace under high purity N2 atmosphere. The weight ratio of NaOH to the pre-oxidized nanofiber were 1, 2, and 4, respectively. The resulting materials were dealt with 0.3 M HCl and distilled water until pH was close to 7.0 at room temperature. The materials were dried under vacuum at 60 °C for 24 h and the final ACFs was obtained, denoted as ACF-1, ACF-2, and ACF-3, respectively.
Preparation of electrode and its capacitive performance evaluation
The electrode was prepared by pressing 70 wt% ACF and 30 wt% PVDF binder into square, which were loaded by 1 × 1 cm2 nickel foam, and dried under vacuum at 60 °C for 24 h afterwards. Cyclic voltammetry (CV), galvanostatic (GV) charge/discharge and electrochemical impedance spectroscopy (EIS) were measured at the room temperature using a CHI 660C electrochemical workstation. The used three-electrode system consisted of a Pt wire as counter electrode, an Hg/Hg2Cl2 electrode as reference electrode, and the deposited sample as working electrode. Nitrogen was bubbled into the electrochemical testing system prior to conducting experiments, with 6 M KOH as electrolyte solution.
Conclusion
In this work, polymerization of AN initiated by AIBN in the presence of Fe(acac)2 presented clear evidence that the polymer chains were growing via RT pattern. The Mn increased and Mw/Mn kept relatively narrow (Mw/Mn < 1.60) as a function of monomer conversion. The diameter distribution (300 nm) of the spinning fibers was homogeneous. The maximum BET surface area of PAN-based ACFs was as high as 1165 m2 g−1. The pore volume of ACF-3 reached 0.14 cm3 g−1. Most is the contribution of the small mesopores of 2–5 nm. The macrostructure of ACFs provided good compatibility with KOH electrolyte. The maximum specific capacitance was as high as 167 F g−1 at ambient temperature. The ACF material also showed well cycle stability after 500 cycles. The maximum energy density was 26.25 W h kg−1. Therefore, ACF material using PAN as the precursor prepared by the novel “living”/controlled method is very suitable and promising electrode materials for EDLCs.
Acknowledgements
This research was supported by the Program for New Century Excellent Talents in University (no. NCET-11-1028), the Natural Science Foundation for Distinguished Young Scholars of Shandong province (no. JQ201203), and the Program for Scientific Research Innovation Team in Universities of Shandong Province.
References
- W. Deng, X. Ji, Q. Chen and C. E. Banks, RSC Adv., 2011, 1, 1171 RSC.
- G. Pognon, T. Brousse and D. Bélanger, Carbon, 2011, 49, 1340 CrossRef CAS PubMed.
- Q. Li, R. Jiang, Y. Dou, Z. Wu, T. Huang, D. Feng, J. Yang, A. Yu and D. Zhao, Carbon, 2011, 49, 1248 CrossRef CAS PubMed.
- B. Xu, F. Wu, R. Chen, G. Cao, S. Chen and Y. Yang, J. Power Sources, 2011, 195, 2118 CrossRef PubMed.
- D. Weingarth, A. Foelske-Schmitz and R. Kötz, J. Power Sources, 2013, 225, 84 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS PubMed.
- M. Khairy and S. A. El-Safty, RSC Adv., 2013, 3, 23801 RSC.
- O. Barbieri, M. Hahn, A. Herzog and R. Kötz, Carbon, 2005, 43, 1303 CrossRef CAS PubMed.
- B. Fang and L. Binder, J. Power Sources, 2006, 163, 616 CrossRef CAS PubMed.
- J. S. Ye, X. Liu, H. F. Cui, W. D. Zhang, F. S. Sheu and T. M. Lim, Electrochem. Commun., 2005, 7, 249 CrossRef CAS PubMed.
- C. Emmenegger, P. Mauron, P. Sudan, P. Wenger, V. Hermann, R. Gallay and A. Züttel, J. Power Sources, 2003, 124, 321 CrossRef CAS.
- B. Xu, F. Wu, S. Chen, C. Zhang, G. Cao and Y. Yang, Electrochim. Acta, 2007, 52, 4595 CrossRef CAS PubMed.
- C. Hu and C. Wang, J. Power Sources, 2004, 125, 299 CrossRef CAS PubMed.
- H. Wang and J. Yao, Ind. Eng. Chem. Res., 2006, 45, 6393 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93 CrossRef CAS PubMed.
- J. Hui, Z. Dong, Y. Shi, Z. Fu and W. Yang, RSC Adv., 2014, 4, 55529 RSC.
- L. Zhou, Z. Zhang, W. Wang, Z. Cheng, N. Zhou, J. Zhu, W. Zhang and X. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 936 CrossRef CAS PubMed.
- Z. Deng, J. Guo, L. Qiu, Y. Zhou, L. Xia and F. Yan, Polym. Chem., 2012, 3, 2436 RSC.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS PubMed.
- D. Liu, H. Chen, P. Yin, N. Ji, G. Zong and R. Qu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2916 CrossRef CAS PubMed.
- Z. Zhang, W. Wang, Z. Cheng, J. Zhu, N. Zhou, Y. Yang, Y. Tu and X. Zhu, Macromolecules, 2010, 43, 7979 CrossRef CAS.
- X. Zhang, W. Wang, K. Guo, C. Wesdemiotis, Z. Zhang and X. Zhu, Polym. Chem., 2013, 4, 637 RSC.
- Y. Wang, Y. Zhang, B. Parker and K. Matyjaszewski, Macromolecules, 2011, 44, 4022 CrossRef CAS.
- T. Guo, L. Zhang, H. Jiang, Z. Zhang, J. Zhu, Z. Cheng and X. Zhu, Polym. Chem., 2011, 2, 2385 RSC.
- Y. Kwak, A. J. Magenau and K. Matyjaszewski, Macromolecules, 2011, 44, 811 CrossRef CAS.
- F. Tang, L. Zhang, J. Zhu, Z. Cheng and X. Zhu, Ind. Eng. Chem. Res., 2009, 48, 6216 CrossRef CAS.
- B. B. Wayland, G. Poszmik, S. L. Mukerjee and M. Fryd, J. Am. Chem. Soc., 1994, 116, 7943 CrossRef CAS.
- A. Debuigne, R. Poli, C. Jérôme, R. Jérôme and C. Detrembleur, Overview of cobalt-mediated radical polymerization: roots, state of the art and future prospects, Prog. Polym. Sci., 2009, 34, 211 CrossRef CAS PubMed.
- Z. Xue, J. C. Daran, Y. Champouret and R. Poli, Inorg. Chem., 2011, 50, 11543 CrossRef CAS PubMed.
- K. Matyjaszewski, Progress in controlled radical polymerization: mechanisms and techniques, ACS, Washington, DC, 2012, ch. 15, pp. 232–240 Search PubMed.
- S. H. Yoon, S. Lim, Y. Song, Y. Ota, W. Qiao, A. Tanaka and I. Mochida, Carbon, 2004, 42, 1723 CrossRef CAS PubMed.
- M. F. Cunningham, Prog. Polym. Sci., 2002, 27, 1039 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS PubMed.
- A. Debuigne, C. Michaux, C. Jérôme, R. Jérôme, R. Poli and C. Detrembleur, Chem. –Eur. J., 2008, 14, 7623 CrossRef CAS PubMed.
- A. K. Gupta and N. Chand, Eur. Polym. J., 1979, 15, 899 CrossRef CAS.
- B. H. Kim, K. S. Yang and H. G. Woo, Electrochem. Commun., 2011, 13, 1042 CrossRef CAS PubMed.
- C. M. Chuang, C. W. Huang, H. Teng and J. M. Ting, Energy Fuels, 2010, 24, 6476 CrossRef CAS.
- J. Tang, D. Q. Liu, Y. X. Zheng, X. W. Li, X. H. Wang and D. Y. He, J. Mater. Chem. A., 2014, 2, 2585 CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.