
Role of propane sultone as an additive to improve the performance of a lithium-rich cathode material at a high potential
Julie Piresa,
Laure Timpermana,
Aurore Castetsb,
Jésus Santos Peñaa,
Erwan Dumontc,
Stéphane Levasseurd,
Rémi Dedryvèreb,
Cécile Tessierc and
Mérièm. Anouti*a
aPCM2E, EA 6299, Université F. Rabelais de Tours, Parc de Grandmont, 37200 Tours, France. E-mail: meriem.anouti@univ-tours.fr; Tel: +33 247366951
bIPREM, CNRS, University of Pau, Helioparc, 2 av. du Prés. Angot, 64053 Pau cedex 09, France
cSAFT, b Saft, 111 Boulevard Alfred Daney, 33074 Bordeaux Cedex, France
dUMICORE, Broekstraat 31, 1000 Brussels, Belgium
First published on 27th April 2015
Abstract
This study presents the use of 1,3-propane sultone (PS) in the [EC–DMC + 1 mol L−1 LiPF6] electrolyte as a protective additive for the Li-rich-NMC xLi2MnO3–(1 − x)LiMO2 (x ≫ 1; M = Ni, Co, Mn) cathode–electrolyte interface during cathode material activation and cycling at a high potential (5 V vs. Li). The results showed that the presence of 1% PS (w/w) ensured complete and better electrode activation during the first cycle than EC–DMC + 1 mol L−1 LiPF6. Thus, Li//Li-rich-NMC half-cell and Gr//Li-rich-NMC full-cell provided capacities as high as C = 330 mA h g−1 during charge and C = 275 mA h g−1 during discharge with a higher cut-off voltage of 5 V. Measurements by cyclic voltammetry demonstrated that activating at such a voltage enhanced the redox activity from Li2MnO3 activation. At same time, the contribution of nickel and cobalt electroactivity is decreased at their regular voltage. This feature was attributed to structural modifications occurring on the surface to the bulk of the material. Long-cycling tests of Li//Li-rich-NMC half-cells with PS provided a higher reversible capacity and superior capacity retention (245 mA h g−1 after 240 cycles) with good coulombic efficiency (99 ± 1%) and better high-discharge rate capability (above 180 mA h g−1 at 1 C regime) than those obtained using conventional electrolytes without additive.
1. Introduction
Although layered Li-rich-NMC cathodes with the specific xLi2MnO3–(1 − x)LiMO2 (x ≫ 1) dual structure may provide remarkably high specific capacities compared to standard NMC,1,2 their practical application is still limited due to unsolved issues.3–5 For instance, transformation from a layered to a spinel-layered intergrowth structure in the activation process during the initial charge leads to oxygen release during the first charge,6 lowering the oxidation state of transition metal ions at the end of the subsequent discharge. This redox process leads to a continuous decay of the discharge plateau by various metal dissolutions and increases the oxidation states of the remaining transition metals at the surface regions of the Li-rich-NMC particles.7 The consequences of these combined phenomena cause accelerated electrolyte decomposition and hence, severely decreased capacity during long-term cycling.8Various strategies have been developed to improve the electrochemical performance of these materials by controlling the particle size within the nanometer scale,9 morphology and composition of interior micro-/nano-structures or by controlling the structural homogeneity and affording a narrow particle-size distribution.10 The major drawbacks of spinel-layered intergrowth formed after activation are its structural instability at a high potential and manganese dissolution into the electrolyte subsequent to the electrode attack by hydrofluoric acid (HF) produced by the reaction of LiPF6 and traces of H2O in the electrolyte.
Notably, using electrolyte additives is a very efficient and economic means of obtaining the desired cell functionality. Adsorbed or solid species on the cathode surface could alleviate the undesirable exothermic reaction between the delithiated cathode and the electrolyte.11 Various additives in the electrolyte are devoted to the formation of a stable SEI on the graphite anode or a protective film on selected cathodes. The best lithium-ion battery performance has been shown by the addition of additives, such as vinylene carbonate (VC),12 ethylene sulfite (ES),13 1,4-butane sultone (BS),14 vinylethylene carbonate,15 and/or lithium bis(oxalate)borate (LiBOB),16 to conventional electrolytes based on alkyl-carbonates. These additives are generally more easily electroactivated than the electrolyte, creating an effective layer during the first cycle and consequently protecting the electrode/electrolyte from decomposition. Although the beneficial effects of these additives on anodes are reasonably well understood, the effects on the Li-rich cathode have rarely been reported. Yang et al. examined the use of tris(hexafluoro-iso-propyl)phosphate (HFiP) as a cathode “SEI” former for a 5 V class spinel LNMO cathode to improve the electrochemical performance of the Li[Li0.2Mn0.56Ni0.16Co0.08]O2 cathode.17 More recently, Choi et al. demonstrated the good effect of LiBOB as an oxidative additive to prevent the decomposition of the electrolyte on the surface of the Li-rich cathode in half-cells and full-cells based on a graphite anode.18 1,3-Propane sultone (PS) is well known to suppress the swelling of batteries at elevated temperatures19 by preventing the electrolyte reaction with oxygen produced by the cathode materials (such as LixCoO2, LixMn2O4, and LixNiO2).20,21 In this study, we used PS as additive to improve the cycling ability of the Li-rich-NMC cathode with a high-limit potential up to 5 V.
2. Materials and methods
All compounds used in the electrolyte formulation were purchased from Sigma Aldrich with purity >99.9% and were used without further purification. Ethylene carbonate (EC) and dimethyl carbonate (DMC) in a binary mixture 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w) with 1 mol L−1 LiPF6 was selected as a baseline electrolyte. 1,3-Propane sultone (PS) additive was added (1% in weight ratio) to this baseline electrolyte. Electrolyte preparation occurred in an argon-filled glove box with oxygen and water contents lower than 1 ppm.
:1 (w/w) with 1 mol L−1 LiPF6 was selected as a baseline electrolyte. 1,3-Propane sultone (PS) additive was added (1% in weight ratio) to this baseline electrolyte. Electrolyte preparation occurred in an argon-filled glove box with oxygen and water contents lower than 1 ppm.
Viscosities were measured using an Anton Parr digital vibrating tube densitometer (model 60/602, Anton Parr, France) and an Anton Parr rolling-ball viscometer (model Lovis 2000 M/ME, Anton Parr, France). A Crison (GLP 31) digital multifrequency conductimeter was used to measure the ionic conductivities. Temperature control from 25 °C to 80 °C was ensured by a JULABO F25 thermostated bath with an accuracy of ±0.2 °C.
Electrochemical properties of the Li-rich-NMC cathode were evaluated with a VMP multichannel potentiostatic–galvanostatic system (Biologic Science Instrument, France) using Swagelok-type cells for the cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements. Galvanostatic cycling was performed on coin-cells under different regimes: C/10, C/5 and C, where C was the courant associated with the theoretical extraction/insertion of one mole of lithium per mole of active material in one hour. All systems were assembled in the glove box. A double microfiber separator drenched with the electrolyte was used for all the tests. Highly pure lithium foils was used as the counter electrode and reference electrode. XPS measurements were carried out with a Kratos Axis Ultra spectrometer, using a focused monochromatized Al Kα radiation, directly connected through a transfer chamber to an argon dry box, in order to avoid moisture/air exposure of the samples. After electrochemical experiments, the positive electrode was carefully separated from other battery components in the argon dry box, washed with DMC solvent to remove the electrolyte, and dried prior to being introduced into the XPS vacuum chamber. Further details can be found elsewhere.22
3. Results and discussion
3.1 Characterization of electrolyte
Fig. 1 shows the conductivity and viscosity of EC–DMC (w/w) + 1 M LiPF6 (red) and EC–DMC (w/w) + 1 M LiPF6 with 1% PS (which corresponded to 5% in molar fraction) as electrolytes from 10 to 80 °C. | ||
| Fig. 1 Evolution of the conductivity, σ and viscosity, η, according to temperature for (EC–DMC) + 1 mol L−1 LiPF6 with 1% PS (blue) and without 1% PS (red). | ||
From this figure, it can be shown that although the viscosity of the mixture with PS was unfavorable at any temperature, the conductivity of the PS-based electrolyte is better. In fact, the Walden product (ση) is more appropriate to compare ion mobility in different electrolytes and Table 1 presents the comparative values at 25 °C and 60 °C for both the electrolytes.
| EC–DMC (w/w) 1 M LiPF6 | EC–DMC (w/w) 1 M LiPF6 + 1% PS | |||
|---|---|---|---|---|
| T | 25 °C | 60 °C | 25 °C | 60 °C |
| σ (mS cm−1) | 12.6 | 20.8 | 13.1 | 22.2 |
| η (mPa s) | 3.26 | 1.77 | 3.68 | 1.99 |
| W (mS mPa cm−1 s) | 41.07 | 36.81 | 48.21 | 43.78 |
The better transport properties observed with 1% of PS (referred to EC–DMC) shown by the Walden values can be explained by the compromise between fluidity in the presence of PS and favorable solvation of the lithium cation by charge distribution in the electron-rich region of the PS ring (inset in Fig. 1), which exceeded that of the EC–DMC binary mixture.
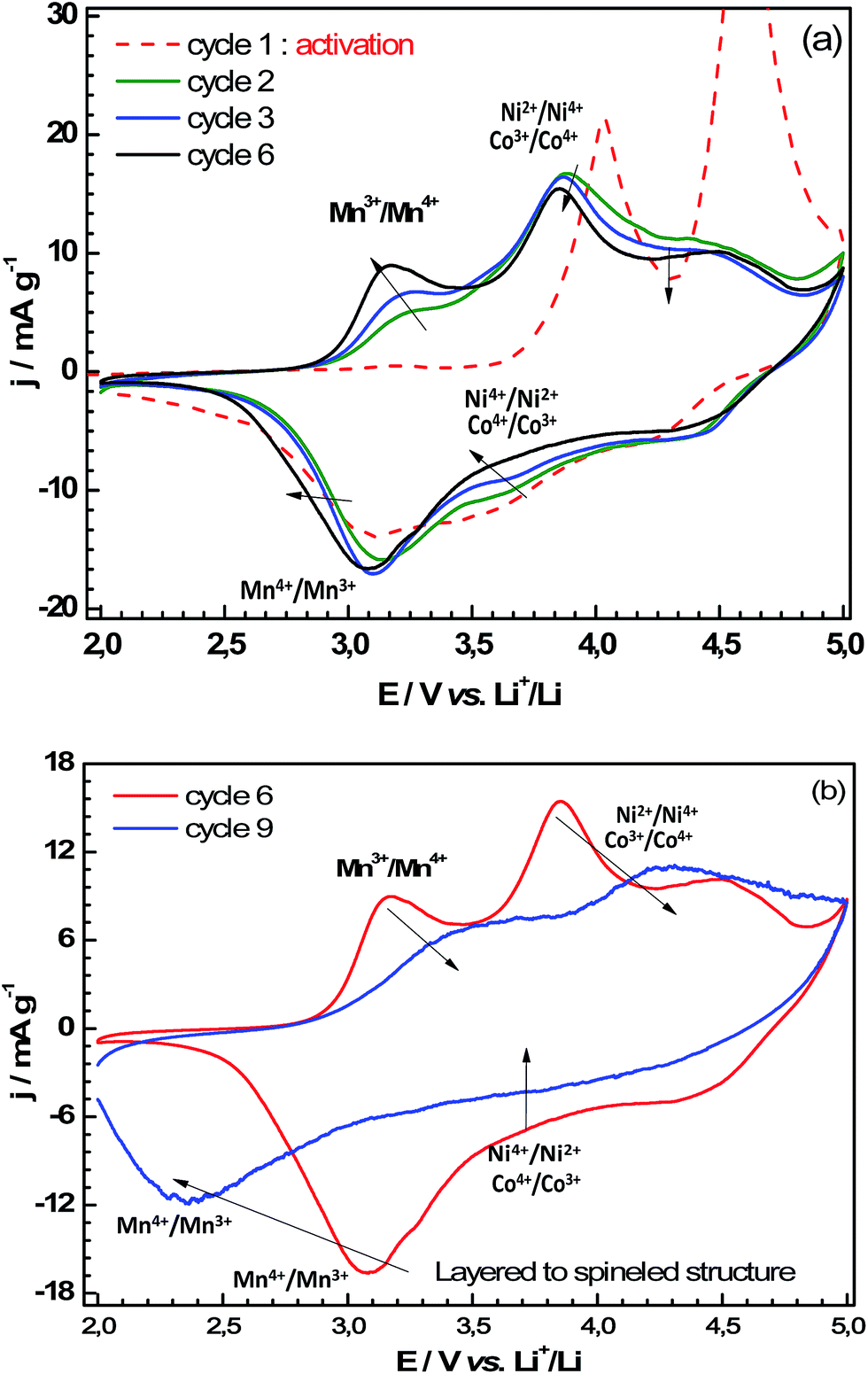 | ||
| Fig. 2 Charge and discharge CV profiles for (a) the first to sixth cycle and (b) the sixth to ninth cycles for the Li-rich-NMC cathode in half Swagelok at 20 μV s−1. | ||
During the subsequent discharge, the anodic curve contained three peaks located close to 4.4 V, 3.7 V and 3.1 V. The partial reversibility of the process responsible for the large charging capacity (oxidation peak at 4.6 V) leads to the reduction peak at 4.4 V in discharge. The peak at 3.7 V corresponds to the reversible reduction of Co4+ to Co3+ and Ni4+ to Ni2+. The peak at 3.1 V corresponds to the reduction of Mn4+ to Mn3+. The last couple was activated upon cycling as the reduction peak at 3.1 V and its oxidation counterpart at 3.2 V become more and more intense at each cycle. Conversely, the electroactivity of nickel and cobalt redox couples decreased upon cycling.
From the sixth cycle (Fig. 2b), a drastic CV profile change occurred. Indeed, the reduction/oxidation peaks are shifted, which corresponds to important kinetic limitations. Important structural modifications of the material could be at the origin of these kinetic limitations, such as the formation of spinel phase due to the very high upper limit voltage (5 V).3
Fig. 3a shows the first charge–discharge profiles of the Li-rich-NMC in EC–DMC without (blue curve) and with 1% PS (red and black curves) in four half-cells at a current density of 16 mA g−1 (0.1 C rate) with different cut-off potentials during charge (4.7 and 5.0 V) and discharge (3 V and 2 V). In the first charge profile, a reproducible wide plateau appeared around the high-potential region (4.4–4.7 V), which was ascribed mainly to the electrochemical activation process of the active material, and whose mechanism is still under debate in the literature.24–26 A shoulder followed this plateau between 4.7 and 5 V, which was assigned to the structural changes detected previously by cyclic voltammetry. This transformation consumed almost 50 mA h g−1 of the total charge capacity (324 mA h g−1).
 | ||
| Fig. 3 Charge and discharge curves at the first cycle for different cut-off charge potentials for half-cell Li//Li-rich-NMC (a), and full-cell Gr//Li-rich-NMC (b) in EC–DMC 1 M LiPF6 + 1% PS at 0.1 C-rate. | ||
Furthermore, activation cut-off potential influenced the initial discharge capacity measured at 2.0 V. Thus, capacity values of 225 mA h g−1 and 275 mA h g−1 (+22%) were provided by the half-cells initially charged at 4.7 V and 5 V, respectively. However, if the discharge was stopped at 3 V (before the plateau relative to Mn4+/Mn3+), although the discharge capacity was also higher for the 5 V-activated system, the relative increase was lower (10% vs. 22%). This proved undoubtedly that activation at 5 V essentially increased the capacity relying on the manganese couple.
Under the same test conditions, full cells with graphite as the negative electrode showed similar charge discharge profiles (Fig. 3b). Although there were no great changes in the charge curves during charging, the shape of the discharge curves changed obviously below 4.7 V, especially with the development of an apparent feature at about 3.0 V.
Fig. 4 shows the temperature effect on the first charge–discharge cycle at the 5 V cut-off potential activation in the presence of 1% PS. The shoulder in the sloping region at 4.8–5.0 V, attributed to the structure change (layered to spinel), was more marked at a high temperature. In fact, it consisted of a sloping region and a pseudo-plateau. Moreover, on discharge, the high-potential plateau close to 3.7 V shifted abruptly to a new feature at 3.0 V, associated with the spinel structure formed from the layered structure. Therefore, such transformation was accelerated at 60 °C.
 | ||
| Fig. 4 Effect of temperature on charge–discharge curves during the first cycle for half-cell Li//Li-rich-NMC in EC–DMC 1 M LiPF6 + 1% PS at 0.1 C rate. | ||
It is known that oxidation voltages as high as 4.7 V lie just above the upper limit of the stability window of alkylcarbonate electrolytes. This leads to deleterious side reactions, which decrease the overall capacity and cycle life of the cathode.27,28 One way to combat this issue is to form a protective film over the active material by coating or by the addition of an additive in the electrolyte. Herein, a film formed by a PS additive through its adsorption or a redox reaction could prevent side reactions and dissolution of metal ions.
The abovementioned observation suggests that charging at high potential (5 V) should lead to a greater transformation of the layered to spinel structure (as the discharge plateau indicated). While this high potential allows for increased energy, it also exacerbates unwanted side reactions between the active cathode material and the electrolyte solution. Addition of PS can lead to the formation of a protective film at the surface, which in turn can achieve more complete activation.
In this study, competition existed at a high potential between structure transformation and manganese dissolution by electrolyte attack and disproportionation of Mn3+, as shown in Fig. 5.
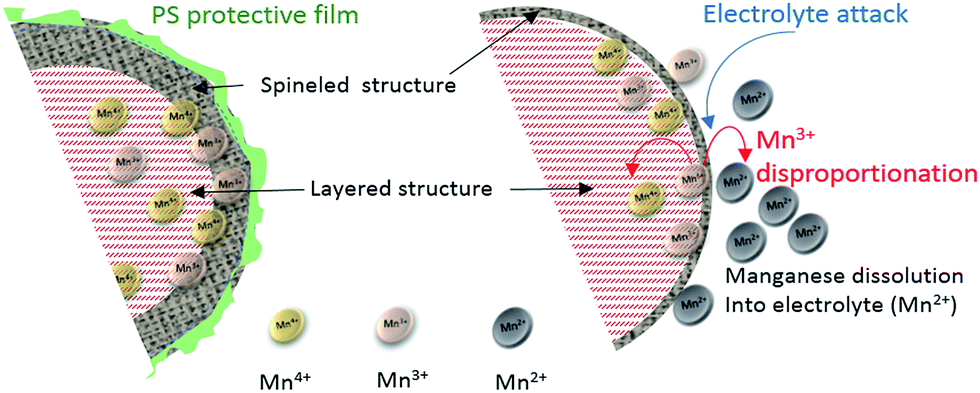 | ||
| Fig. 5 Schematic of the PS protection mechanism on the Li-rich-NMC cathode during cycling and progressive transformation from the layered to the spinel structure. | ||
In the case of a PS-protected interface, the bulk-layered structure to spinel transformation was preserved without the inconvenience of electrode attack by HF or the exposure to the electrolyte of Mn3+, which is prone to disproportion. Therefore, manganese dissolution was mitigated and the relative stability of this new spinel during the cycling was high. This was consistent with the maintenance of the manganese plateau observed during the discharge (Fig. 5, left), while the non-protected layered structure in the standard electrolyte shrank fast leading to dissolution of transition metal ions (Fig. 5, right).29,30
In summary, the addition of PS contributed to the formation of a protective film on the particle surface with the main role of preserving the integrity of the spinel formed at high voltage by protecting the particles from electrolyte erosion and preventing the dissolution of transition metal ions from the bulk material.
 | ||
| Fig. 6 Electrochemical impedance spectroscopy plots of Li-rich-NMC charged at 4.6 V, 4.7 V and 5 V vs. Li+/Li recorded in the three electrode cells at 25 °C in EC–DMC 1 M LiPF6 + 1% PS. Dots and lines correspond to experimental and fitted results, respectively. | ||
The presence of an adsorbed film (instead of a solid film) on the electrode surface was consistent with the stability of the electrolyte components up to 4.7 V. Interestingly, the charge transfer resistance (from 205 Ω to 240 Ω) after traditional activation, indicates good electrode/electrolyte interfacial properties for this cathode/electrolyte couple. However, when charged up to 5.0 V, the Nyquist plots could not be fitted with the circuit described above. Instead, a Randles classic circuit containing the Rint//Cint element and the two other R//C elements associated with two solid films can describe the interfacial issues.
Therefore, raising the potential to 5 V displaced the adsorbed film and created two solid interfaces on the electrode. These solid films may be formed by the degradation of PS at such high potential and can consist of sulfur-containing derivatives. Furthermore, independently on these resistive films, a huge increase of the Rct (from 240 Ω to 1300 Ω) was shown, which is consistent with deep reorganization of the system at 5 V that worsens the electrode electronic properties. This latter characteristic is in agreement with the mechanism proposed above.
 | ||
| Fig. 7 O 1s XPS spectra of positive electrodes before cycling (starting electrode) and charged up to 4.6 V, 4.7 V and 5 V vs. Li+/Li with EC–DMC + 1% PS + 1 M LiPF6. | ||
 | ||
| Fig. 8 Discharge capacity and efficiency data as a function of cycle number at different scan rates (a) and energy (b) of Li-rich-NMC EC–DMC 1 M LiPF6 + 1% PS in the 2.0–5.0 V potential range at 25 °C. | ||
4. Conclusions
Based on the experimental results of this study, a number of conclusions were derived. First, we showed that propane sultone was an efficient additive in EC–DMC 1 M LiPF6 to form a protective film on Li-rich-NMC with a wide electrochemical stability window (5.0 V). It is plausible that a thin layer of PS on Li-rich-NMC particles could provide the necessary protection of the outer surface material from electrolyte attack and prevent side reactions and dissolution of metal ions. Secondly, this protection allowed further activation at a potential higher than the conventional 4.6 V proposed for Li-rich-NMC materials. This “new” activation step at 5 V probably led to the transformation of the bulk layered structure to a spinel phase with substantial manganese rearrangement, resulting in accessory manganese redox activity at voltages lower than 3.0 V. Transformation was also strongly affected by temperature and controlled by the initial charge–discharge cut-off voltage. We showed by cyclic voltammetry, galvanostatic measurements and EIS that the continuous increase of the stable manganese in a spinel structure at the surface could protect the cathode from etching by the HF generated in the electrolyte at a high potential. This protection suppresses the degradation of the cathode and restrains the dissolution of transition metal ions from the bulk material, and thus improves the long-term cycling stability of the cathode but does not prevent the midpoint voltages fading.Acknowledgements
This research was supported by “Agence National de la recherche” through the “Alisé” project.Notes and references
- H. Yu and H. Zhou, J. Phys. Chem. Lett., 2013, 4, 1268–1280 CrossRef CAS.
- F. Cheng, J. Liang, Z. Tao and J. Chen, Adv. Mater., 2011, 23, 1695–1715 CrossRef CAS PubMed.
- H. Yu, H. Kim, Y. Wang, P. He, D. Asakura, Y. Nakamura and H. Zhou, Phys. Chem. Chem. Phys., 2012, 14, 6584–6595 RSC.
- S. Qiu, Z. Chen, F. Pei, F. Wu, Y. Wu, X. Ai, H. Yang and Y. Cao, Eur. J. Inorg. Chem., 2013, 2013, 2887–2892 CrossRef CAS PubMed.
- X. Yu, Y. Lyu, L. Gu, H. Wu, S.-M. Bak, Y. Zhou, K. Amine, S. N. Ehrlich, H. Li, K.-W. Nam and X.-Q. Yang, Adv. Energy Mater., 2014, 4, 1300950–1300961 Search PubMed.
- Y. Gao, J. Ma, X. Wang, X. Lu, Y. Bai, Z. Wang and L. Chen, J. Mater. Chem. A, 2014, 2, 4811–4818 CAS.
- S. K. Martha, J. Nanda, Y. Kim, R. R. Unocic, S. Pannala and N. J. Dudney, J. Mater. Chem. A, 2013, 1, 5587–5595 CAS.
- X. Yang, D. Wang, R. Yu, Y. Bai, H. Shu, L. Ge, H. Guo, Q. Wei, L. Liu and X. Wang, J. Mater. Chem. A, 2014, 2, 3899–3911 CAS.
- A. Boulineau, L. Simonin, J.-F. Colin, C. Bourbon and S. Patoux, Nano Lett., 2013, 13, 3857–3863 CrossRef CAS PubMed.
- L. Zhang, B. Wu, N. Li, D. Mu, C. Zhang and F. Wu, J. Power Sources, 2013, 240, 644–652 CrossRef CAS PubMed.
- L. Zhang, W. Borong, L. Ning and W. Feng, Electrochim. Acta, 2014, 118, 67–74 CrossRef CAS PubMed.
- D. Aurbach, K. Gamolsky, B. Markovsky, Y. Gofer, M. Schmidt and U. Heider, Electrochim. Acta, 2002, 47, 1423–1439 CrossRef CAS.
- M. Lu, H. Cheng and Y. Yang, Electrochim. Acta, 2008, 53, 3539–3546 CrossRef CAS PubMed.
- M. Q. Xu, W. S. Li, X. X. Zuo, J. S. Liu and X. Xu, J. Power Sources, 2007, 174, 705–710 CrossRef CAS PubMed.
- J. Li, W. Yao, Y. S. Meng and Y. Yang, J. Phys. Chem. C, 2008, 112, 12550–12556 CAS.
- E. P. Roth, D. H. Doughty and J. Franklin, J. Power Sources, 2004, 134, 222–234 CrossRef CAS PubMed.
- S. Tan, Z. Zhang, Y. Li, Y. Li, J. Zheng, Z. Zhou and Y. Yang, J. Electrochem. Soc., 2013, 160, A285–A292 CrossRef CAS PubMed.
- S. J. Lee, J.-G. Han, I. Park, J. Song, J. Cho, J.-S. Kim and N.-S. Choi, J. Electrochem. Soc., 2014, 161, A2012–A2019 CrossRef CAS PubMed.
- H. Lee, S. Choi, S. Choi, H.-J. Kim, Y. Choi, S. Yoon and J.-J. Cho, Electrochem. Commun., 2007, 9, 801–806 CrossRef CAS PubMed.
- U. von Sacken, E. Nodwell, A. Sundher and J. R. Dahn, J. Power Sources, 1995, 54, 240–245 CrossRef CAS.
- K.-K. Lee, W.-S. Yoon, K.-B. Kim, K.-Y. Lee and S.-T. Hong, J. Electrochem. Soc., 2001, 148, A716–A722 CrossRef CAS PubMed.
- L. El Ouatani, R. Dedryvère, C. Siret, P. Biensan, S. Reynaud, P. Iratçabal and D. Gonbeau, J. Electrochem. Soc., 2009, 156, A103–A113 CrossRef CAS PubMed.
- A. R. Armstrong, M. Holzapfel, P. Novák, C. S. Johnson, S.-H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS PubMed.
- M. M. Thackeray, S.-H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112–3125 RSC.
- S. Hy, F. Felix, J. Rick, W.-N. Su and B. J. Hwang, J. Am. Chem. Soc., 2014, 136, 999–1007 CrossRef CAS PubMed.
- M. Sathiya, A. M. Abakumov, D. Foix, G. Rousse, K. Ramesha, M. Saubanère, M. L. Doublet, H. Vezin, C. P. Laisa, A. S. Prakash, D. Gonbeau, G. VanTendeloo and J. M. Tarascon, Nat. Mater., 2015, 14, 230–238 CrossRef CAS PubMed.
- S. A. Freunberger, Y. Chen, Z. Peng, J. M. Griffin, L. J. Hardwick, F. Bardé, P. Novák and P. G. Bruce, J. Am. Chem. Soc., 2011, 133, 8040–8047 CrossRef CAS PubMed.
- G. BrucePeter, A. FreunbergerStefan, J. HardwickLaurence and M. TarasconJean, Nat. Mater., 2012, 11, 172–172 Search PubMed.
- D. Tang, L. Ben, Y. Sun, B. Chen, Z. Yang, L. Gu and X. Huang, J. Mater. Chem. A, 2014, 2, 14519–14527 CAS.
- A. Manthiram, K. Chemelewski and E.-S. Lee, Energy Environ. Sci., 2014, 7, 1339–1350 CAS.
- P. Oh, S. Myeong, W. Cho, M.-J. Lee, M. Ko, H. Y. Jeong and J. Cho, Nano Lett., 2014, 14, 5965–5972 CrossRef CAS PubMed.
- J. Zhang, J. Wang, J. Yang and Y. NuLi, Electrochim. Acta, 2014, 117, 99–104 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |