
The importance of Au⋯π(aryl) interactions in the formation of spherical aggregates in binuclear phosphane gold(i) complexes of a bipodal thiocarbamate dianion: a combined crystallographic and computational study, and anti-microbial activity†
Abstract
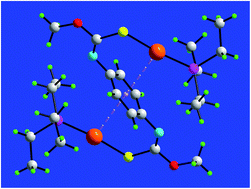
Binuclear phosphanegold(I) complexes of a bipodal thiocarbamate dianion, (R3PAu)2L, R = Et (1), Ph (2) and Cy (3), where LH2 is {1,4-[MeOC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)N(H)]2C6H4}, have been synthesised, and characterised spectroscopically (NMR and IR) and by X-ray crystallography. The gold atoms are linearly coordinated within a P-,S-donor set, and are oriented toward the central ring to form intramolecular Au⋯π(aryl) interactions, rather than the intramolecular Au⋯O interactions normally observed in mononuclear analogues. This phenomenon has been investigated by theory (LC-ωPBE-XDM) for 1 which revealed that the geometry optimised species with two Au⋯π(aryl) interactions is more stable by at least 12 kcal mol−1 compared to conformations having one or more Au⋯O interactions instead. The disk diffusion, minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) methods were used to observe the inhibitory effect of complexes 1–3. The disk diffusion results demonstrated that 1 exhibited a broad spectrum of anti-bacterial activity toward 24 strains of Gram-positive and Gram-negative bacteria. By contrast, the anti-bacterial activity of 2 and 3 was limited to Gram-positive bacteria. Further evaluation showed that 1 exhibited marked bactericidal activity against B. cereus, B. subtilis, E. faecalis, L. monocytogenes, S. aureus, S. saprophyticus and methicillin resistant S. aureus cf. standard antibiotics tetracycline and chloramphenicol.
S)N(H)]2C6H4}, have been synthesised, and characterised spectroscopically (NMR and IR) and by X-ray crystallography. The gold atoms are linearly coordinated within a P-,S-donor set, and are oriented toward the central ring to form intramolecular Au⋯π(aryl) interactions, rather than the intramolecular Au⋯O interactions normally observed in mononuclear analogues. This phenomenon has been investigated by theory (LC-ωPBE-XDM) for 1 which revealed that the geometry optimised species with two Au⋯π(aryl) interactions is more stable by at least 12 kcal mol−1 compared to conformations having one or more Au⋯O interactions instead. The disk diffusion, minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) methods were used to observe the inhibitory effect of complexes 1–3. The disk diffusion results demonstrated that 1 exhibited a broad spectrum of anti-bacterial activity toward 24 strains of Gram-positive and Gram-negative bacteria. By contrast, the anti-bacterial activity of 2 and 3 was limited to Gram-positive bacteria. Further evaluation showed that 1 exhibited marked bactericidal activity against B. cereus, B. subtilis, E. faecalis, L. monocytogenes, S. aureus, S. saprophyticus and methicillin resistant S. aureus cf. standard antibiotics tetracycline and chloramphenicol.
 Please wait while we load your content...
Please wait while we load your content...

