DOI:
10.1039/C5RA05576H
(Paper)
RSC Adv., 2015,
5, 52314-52319
Analysis of chondroitin sulfate from different sources of cartilage by electrophoretically mediated microanalysis†
Received
29th March 2015
, Accepted 4th June 2015
First published on 5th June 2015
Abstract
An electrophoretically mediated microanalysis (EMMA) protocol for the determination of different chondroitin sulfate (CS) origins based on the difference in the content of unsaturated disaccharides produced by degradation with chondroitinase ABC was developed. Separations were performed in an uncoated fused silica capillary (total length: 60.2 cm, effective length: 50 cm, 50 μm i.d.) at 20 kV and 37 °C. The influences of various parameters, such as different kinds of separation buffers, substrate concentration and incubation time, on separation were investigated. The optimum conditions were as follows: separation buffer, 25 mM tetraborate buffer (pH 9.5); incubation buffer, 50 mM Tris-60 mM acetate buffer (pH 8.0); sample injection, 5 s at 0.5 psi; CS concentration, 500 μg mL−1; incubation time, 8 min. Nonsulfated, monosulfated, disulfated and trisulfated Δ-disaccharides were separated well under the above optimal conditions. The developed method was used to determine the amounts of disaccharides in CS from different sources and the results were compared with those obtained by offline analysis. The results indicated that the developed method could successfully distinguish CS with minor differences and could obtain good coherence results compared with the traditional method.
1 Introduction
Chondroitin sulfate (CS) is one kind of anionic biological macromolecules with the property of microheterogeneity and a wide variety of physiological functions.1 It is often used to treat osteoarthritis (OA) by oral administration combined with glucosamine (GlcN),2 sometimes it is used for treating ophthalmologic diseases.3 It has been also reported that CS may help in the treatment of psoriasis.3 CS consists of an alternate sequence of D-glucuronic acid (GlcA) and aminohexose linked by β(1 → 3) bonds and there are eight kinds of disaccharides according to the position and extent of sulfation (Fig. 1). CS is widespread in the extracellular matrix of cartilage and other connective tissues mainly sourced from cows, fowls, pigs and sharks,4 and is widely used as a nutraceutical and pharmaceutical raw material. Because CS from different origins may have microheterogeneity in its structure including ratios of various sulfated disaccharides, sites and degrees of sulfation and molecular mass, which finally provide different physiological functions,5,6 it is important to identify the composition of disaccharides of CS from different biological tissue.
 |
| | Fig. 1 Repeat unit of chondroitin sulfate. R1 = R2 = R3 = H, ΔUA → GalNAc Na; R1 = SO3− and R2 = R3 = H, ΔUA → GalNAc-4S Na2, CSA; R2 = SO3− and R2 = R3 = H, ΔUA → GalNAc-6S Na2, CSC; R2 = R3 = SO3− and R1 = H, ΔUA-2S → GalNAc-6S Na3, CSD; R1 = R2 = SO3− and R3 = H, ΔUA → GalNAc-4S-6S Na3, CSE; R1 = R3 = SO3− and R2 = H, ΔUA-2S → GalNAc-4S Na3, CSB; R1 = R2 = R3 = SO3−, ΔUA-2S → GalNAc-4S-6S Na4; R1 = R2 = H and R3 = SO3−, ΔUA-2S → GalNAc Na2. | |
At present, the techniques applied for the determination of different CS sources involve nuclear magnetic resonance (NMR) spectroscopy,7,8 Fourier transform infrared (FTIR) spectroscopy,9 near infrared spectroscopy (NIRS),10 strong ion-exchanged HPLC (high performance liquid chromatography) and CE (capillary electrophoresis) method.11 However, NMR is too expensive for routine analysis of CS origin, FTIR demands a high level of sample treatment skill, NIRS needs a large amount of samples and complex data processing, and ion-exchange HPLC requires an expensive column. In contrast, CE is an optional technique, but in general, it still needs offline degradation before online detection. In most cases, enzymes are expensive and the degradation process is laborious. Since CE is a separation technique with the advantage of a small quantity of sample and free solution analysis, an analytical protocol based on CE analysis, electrophoretically mediated microanalysis (EMMA), emerged. For EMMA, both the enzymatic reaction and the separation of the reactants and products can take place in the capillary. Enzyme and substrate can be mixed in the CE capillary based on the differences in their electrophoresis velocity. EMMA is a more automatic and economic technique, but to date few work about CS in-capillary enzyme reaction has been reported. Hitoshi Okamoto et al. employed an EMMA method for the analysis of CS.12 However, the report focused on the determination of the CS via degradation by four kinds of enzymes, and the results are far from enough for the determination of CS sources because of the separation of the disaccharides was insufficient.12 Thus developing an accurate, efficient and convenient analytical method for determining the origin of CS, even each tissue type CS of one kind of animal, is meaningful.
Since CS from different sources can produce various kinds and ratios of disaccharides after degradation,11,13 we aimed to develop an EMMA method to identify the source of CS in this study. To achieve this goal, an EMMA method employing both partial filling and a voltage switch sequence was optimized with the consideration of resolution, sensitivity, and reproducibility. The EMMA method can be used for the discrimination of CS with minor differences.
2 Materials and methods
2.1 Chemicals and reagents
CS reference standard substance from porcine cartilage was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). CS from bovine cartilage was donated by Zaozhuang Sainuokang Biochemistry Co. (Zaozhuang, China). CS from shark cartilage and chondroitinase ABC were purchased from Sigma-Aldrich (St. Louis, MO, USA). Eight standard disaccharides (sodium salts) were from Dextra (Reading, UK): ΔUA → GalNAc Na (ΔDi-0S); ΔUA → GalNAc-4S Na2 (ΔDi-4S); ΔUA → GalNAc-6S Na2 (ΔDi-6S); ΔUA → GalNAc-4S,6S Na3 (ΔDi-diSE); ΔUA-2S → GalNAc-4S Na2 (ΔDi-diSB); ΔUA-2S → GalNAc-6S Na3 (ΔDi-diSD); ΔUA-2S → GalNAc-4S,6S Na4 (ΔDi-triS); ΔUA-2S → GalNAc Na2 (ΔDi-UA2S). Tris(hydroxymethyl)aminomethane (99%) and sodium acetate trihydrate (A.R.) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Hydrochloric acid (A.R.) was purchased from Laiyang Fine Chemical Factory (Laiyang, China). Sodium tetraborate, sodium hydroxide, sodium phosphate monobasic dihydrate, and phosphoric acid (85%) were of analytical grade and supplied by Tianjin Guangcheng Chemical Factory (Tianjin, China). Deionized water was obtained from a Millipore Milli-Q Biocell purification system (Bedford, America).
2.2 Instrument and conditions
CE was performed on a PA 800 plus capillary electrophoresis system (Beckman Coulter, Fullerton, CA) equipped with an autosampler, a PDA detector and a temperature control system (15–60 °C ± 1 °C). Samples were introduced by pressure injection at 0.5 psi for 5 s from the anode and detected at the cathode under alkaline conditions and the pH of BGE was adjusted by pH meter from Sartorius (Gottingen, Germany). Fused-silica capillary of 50 μm i.d., 375 μm o.d. with a total length of 60.2 cm (50 cm effective length) was from Yongnian Optical Fiber Factory (Baoding, China). The new capillary was first rinsed at 20 psi using the following reagents in sequence: water purified with a Milli-Q system for 5 min, 0.1 M NaOH solution for 30 min, purified water for 5 min followed by the running buffer for 10 min, and then kept for 5 min under 20 kV. Between runs, the capillary was washed at 20 psi with purified water for 2 min, 0.1 M NaOH solution for 3 min, purified water for 2 min and fresh BGE for 3 min. The samples were maintained at 4 °C and the capillary was at 37 °C by liquid cooling. The detection wavelength was 232 nm for disaccharides. Electropherograms were obtained and analyzed using 32 Karat 9.1 software (Beckman Coulter).
2.3 Preparation of reagents and samples
The separation buffer (pH 9.5, 25 mM tetraborate buffer) was prepared by dissolving sodium tetraborate with purified water into the desired concentration and the pH was adjusted with 1 M NaOH solution. Phosphate buffer was prepared by dissolving sodium phosphate monobasic dehydrate with purified water into the desired concentration and pH was adjusted with 50% phosphoric acid solution. Tris phosphate buffer was prepared by dissolving Tris with purified water into the desired concentration and pH was adjusted with 50% phosphoric acid solution. The incubation buffer (pH 8.0, 50 mM Tris-60 mM sodium acetate buffer) was prepared by dissolving Tris and sodium acetate trihydrate into desired concentration with purified water and the pH was adjusted with 50% hydrochloric acid solution. The eight standard disaccharides mixture was prepared in purified water at a final concentration of 125 μg mL−1 for ΔDi-4S, ΔDi-6S and ΔDi-0S and 25 μg mL−1 for ΔDi-triS, ΔDi-diSB, ΔDi-diSD, ΔDi-diSE and ΔDi-UA2S. Different concentrations of CS standard solutions from shark in range of 100–700 μg mL−1 were prepared in incubation buffer. Stock solution of 10 mg mL−1 CS from different origins were prepared with incubation buffer and stored at −80 °C. Different concentrations of standard solutions were obtained by diluting the corresponding stock solutions to desired concentrations with incubation buffer. The samples for identification of disaccharides peaks were prepared as follows: the CS and disaccharide mixture was prepared by adding 10 μL of certain disaccharide standard solution to 100 μL 500 μg mL−1 CS sample, the CS contrast sample was prepared by adding 10 μL of purified water to 100 μL 500 μg mL−1 CS sample. Chondroitinase ABC was prepared at a final concentration of 1 U mL−1 with incubation buffer and dispensed into 1 mL vials and stored at −80 °C. Before daily analysis one vial was taken out to place to room temperature. Prior to analysis, all of the solutions were filtered through 0.22 μm cellulose acetate membrane filters.
2.4 Procedure for offline enzyme reaction analysis
Fifty microliter of the CS solution from different sources (2 mg mL−1) was placed in a 1.5 mL vail, after that 50 μL of chondroitinase ABC solution (1 U mL−1) was added to the solution and finally 100 μL of incubation buffer was added into the mixture. The reaction was allowed to react at 37 °C for 1 h. A portion of the solution was analyzed by CE using the conditions described above and the results were shown in Table 1.
Table 1 Percentages of disaccharides of bovine chondroitin sulfate (BCS), porcine chondroitin sulfate (PCS), chondroitin sulfate from shark (SCS) evaluated by EMMA method (A) and offline method (B)a
| |
BCS |
PCS |
SCS |
| A |
B |
A |
B |
A |
B |
| N.D., not detected. |
| ΔUA → GalNAc Na |
1.690% |
1.870% |
1.890% |
3.120% |
N.D. |
N.D. |
| ΔUA → GalNAc-6S Na2 |
22.22% |
24.33% |
15.13% |
17.33% |
4.600% |
3.390% |
| ΔUA → GalNAc-4S Na2 |
74.45% |
73.80% |
81.51% |
79.55% |
87.48% |
91.08% |
| ΔUA → GalNAc-4S,6S Na3 |
1.640% |
N.D. |
1.470% |
N.D. |
7.920% |
5.530% |
| ΔUA-2S → GalNAc-4S Na3 |
N.D. |
N.D. |
N.D. |
N.D. |
N.D. |
N.D. |
| ΔUA-2S → GalNAc-6S Na3 |
N.D. |
N.D. |
N.D. |
N.D. |
N.D. |
N.D. |
| ΔUA-2S → GalNAc-4S-6S Na4 |
N.D. |
N.D. |
N.D. |
N.D. |
N.D. |
N.D. |
| ΔUA-2S → GalNAc Na2 |
N.D. |
N.D. |
N.D. |
N.D. |
N.D. |
N.D. |
| SO3−/COO− |
0.9993 |
0.9813 |
0.9974 |
0.9688 |
1.079 |
1.055 |
2.5 Procedure of electrophoretically mediated microanalysis (EMMA)
An illustration of the conversion of CS to disaccharide is shown in Fig. 2. Firstly, a plug of incubation buffer (0.5 psi × 10 s) was introduced to the capillary; then, the CS solution (500 μg mL−1, 0.5 psi × 5 s) and enzyme solution (1 U mL−1, 0.5 psi × 5 s) were introduced respectively; after that, another plug of incubation buffer (0.5 psi × 10 s) was introduced; finally, a plug of separation buffer (0.5 psi × 5 s) was injected. After the injection of the enzyme solution, the inlet end of the capillary was allowed to dip into the incubation buffer for 6 s to prevent cross contamination. The mixing was achieved by using a voltage switch sequence: −1 kV/+1 kV/−1 kV/+1 kV, each for 6 s, and then the incubation was kept for 8 min at 1 kV, finally the separation was performed using the same separation conditions as the offline analysis. Relative high concentration of enzyme was employed to insure the complete degradation in short time. The analytical performances of the CE separation were assessed by means of calculating the reproducibility of migration time and peak area. The reproducibility was evaluated by six repeated injections of a standard disaccharides mixture.
 |
| | Fig. 2 Schematic representation of the conversion of CS to disaccharide in the established EMMA method. | |
2.6 Analysis of real samples
The developed method was used for analysis of the CS samples from sharks, pigs and cows. Samples were prepared at a concentration of 500 μg mL−1 with incubation buffer. The four contrast samples containing incubation buffer, chondroitinase ABC and two concentration levels of CS samples were subjected to CE analysis for the identification of the peaks in EMMA electropherogram. Since the level of disaccharides released from CS after enzymatic degradation corresponds to its content in the native glycosaminoglycan, the identification of CS origin was carried out by comparing the differences in the kinds and peak area ratios of disaccharides.
3 Results and discussion
3.1 CZE (capillary zone electrophoresis) separation of the CS disaccharides standard references
Phosphate buffer, Tris phosphate buffer and tetraborate buffer were investigated for the separation of the standard disaccharides mixture and the obtained electropherograms are shown in Fig. 3. Phosphate buffer offered a good separation while the baseline was distorted (Fig. 3A). Tris phosphate buffer could separate eight disaccharides with a steady baseline but it also separated the anomeric forms of ΔDi-UA2S, ΔDi-6S and ΔDi-4S (Fig. 3B) as previously reported.14 Application of high concentration buffer could lead to comigration of the anomers but the width of peak significantly increased due to the high Joule heating, the results are showed in ESI.† By using tetraborate buffer, a satisfied separation as well as a steady baseline was achieved (Fig. 3C). Moreover, the disaccharides and tetraborate could form a complex which significantly enhanced the UV absorption. Further optimization of pH and concentration was carried out and finally pH 9.5, 25 mM tetraborate buffer was chosen as separation buffer. The effect of different voltages on separation was investigated and finally the analysis was conducted at +20 kV.
 |
| | Fig. 3 Electropherogram of a standard mixture of chondro-disaccharides. Concentrations: ΔDi-triS, ΔDi-diSD, ΔDi-diSB, ΔDi-diSE, ΔDi-UA2S: 25 μg mL−1; ΔDi-6S, ΔDi-4S, ΔDi-0S: 125 μg mL−1. Fused silica capillary (total length: 60.2 cm, effective length: 50 cm, 50 μm i.d.). Capillary temperature: 37 °C. Sample temperature: 4 °C. Sample injection: pressure injection at 0.5 psi × 5 s. Detection wavelength: 232 nm. Peaks identification: (1) ΔDi-triS, (2) ΔDi-diSD, (3) ΔDi-diSB, (4) ΔDi-diSE, (5) ΔDi-UA2S, (6) ΔDi-6S, (7) ΔDi-4S, (8) ΔDi-0S. (A) BGE: pH 3.5, 130 mM phosphate buffer. Voltage: −30 kV. (B) BGE: pH 3.0, 170 mM Tris phosphate buffer. Voltage: −30 kV. (C) BGE: pH 9.5, 25 mM tetraborate buffer. Voltage: +20 kV. The inset over (C) magnifies (C) from 4.5 min to 5.4 min. | |
The precision of method was evaluated via the relative standard deviation (RSD) of six repetition runs. The RSDs of migration times and peaks areas for eight disaccharides are all lower than 1.4% and 1.8%, respectively. The results indicated that the developed method had a good reproducibility. The relative data are showed in ESI.†
3.2 Development of the EMMA method
In offline assays, the incubation of CS with chondroitinase ABC was performed at pH 8.0,9,11,13,15,16 while the separation of the unsaturated disaccharides obtained from enzyme degradation was performed at a pH different from its optimal incubation pH. Due to the challenge above, the partial filling method17 was chosen for the development of an in-capillary assay separating the enzyme and CS solutions from BGE by pressure injecting (0.5 psi × 10 s) two plugs of incubation buffer at both sides of the sample zone, as shown in Fig. 2. Another challenge in in-capillary assay is the efficient mixing of enzyme and substrate solutions, which can be achieved by employment of a voltage switch sequence.18,19
The electropherograms of contrast samples indicated that under the optimum conditions enzyme migrated faster than CS (Fig. 4), thus CS was first introduced to the capillary followed by the enzyme. Under this injection principle, the enzyme would traverse across the CS zone under the electric field and initiated the degradation. In addition, a sequence of traverse voltages could make the mixing more thoroughly to ensure a fast and efficient degradation reaction. The optimization of the EMMA conditions was conducted. At first, five levels of CS concentrations (300 μg mL−1, 400 μg mL−1, 500 μg mL−1, 600 μg mL−1 and 700 μg mL−1) were injected into the capillary and analyzed. Take sensitivity and sample consumption into consideration, 500 μg mL−1 was chosen as the optimal concentration, the results are showed in ESI.† The incubation time was investigated at five levels (1 min, 3 min, 5 min, 8 min and 10 min) and the results are shown in Fig. 5. With the incubation time increase from 8 min to 10 min, the peak areas of disaccharides slightly decreased, so 8 min was chosen as the incubation time. The assigned disaccharide peaks were identified by comparing the electropherograms of CS and disaccharides mixtures with CS contrast samples. The results are shown in Fig. 6. The RSDs for the disaccharides were lower than 1.2% for migration time and lower than 1.9% for peak area, which indicated the high reproducibility of the EMMA method. The results are showed in ESI.†
 |
| | Fig. 4 Electropherograms of (A) reaction solution of 100 μg mL−1 CS solution and 1 U mL−1 enzyme solution, (B) incubation buffer, (C) 1 mg mL−1 CS solution, (D) 500 μg mL−1 CS solution, and (E) 1 U mL−1 enzyme solution. Detection wavelength: 200 nm. (1) Solvent peak of incubation buffer, (2) enzyme, (3) ΔDi-6S, (4) ΔDi-4S, (5) CS, (6) solvent peak of incubation buffer. Other conditions were as in Fig. 3C. | |
 |
| | Fig. 5 Effect of the incubation time on the ΔDi-4S production. | |
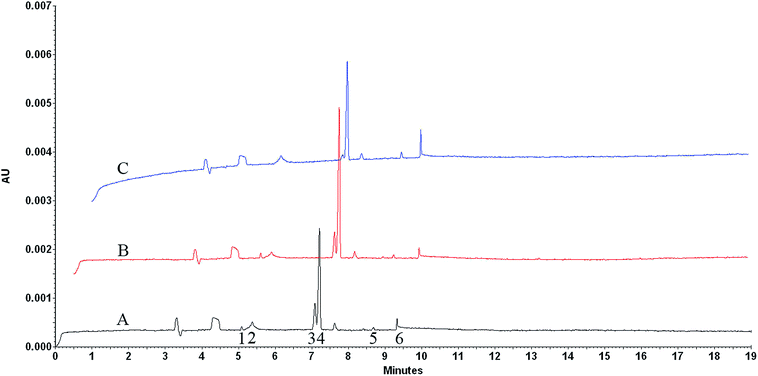 |
| | Fig. 6 Electrophoretically mediated microanalysis of CS from different sources. (A) Cow, (B) pig, (C) shark. Fused silica capillary (total length: 60.2 cm, effective length: 50 cm, i.d. 50 μm). Capillary temperature: 37 °C. Sample temperature: 4 °C. Separation buffer: pH 9.5, 25 mM tetraborate buffer. Incubation buffer: pH 8.0, 50 mM Tris-60 mM acetate buffer (0.5 psi × 10 s). Voltage: +20 kV. Injection: CS solution (0.5 psi × 5 s), enzyme solution (0.5 psi × 5 s). Voltage switch sequence: −1 kV/+1 kV/−1 kV/+1 kV, each for 6 s. In capillary incubation time: 8 min. Detection wavelength: 232 nm. Peaks identification: (1) ΔDi-0S, (2) enzyme, (3) ΔDi-6S, (4) ΔDi-4S, (5) ΔDi-UA2S, (6) solvent peak of incubation buffer. | |
The possibility of the established method for the quantification of CS from shark in the concentration range of 100 μg mL−1 to 700 μg mL−1 was also investigated in this study, while the concentration range of CS in ref. 12 was from 25 μg mL−1 to 125 μg mL−1 by employing four kinds of enzyme.12 In particular, ΔDi-4S was selected for quantitation because of its abundance in samples and short migration time. The analysis was performed in triplicate for each concentration level under the optimized CE conditions and the average peak area of ΔDi-4S was plotted versus the concentration (μg mL−1) of the analyte to obtain the calibration curve. The regression equation was y = 12.10x + 892.14, r = 0.9977. The limit of detection, observed as a peak with a signal-to noise ratio of 3, was found to be 2.0 μg mL−1 of CS solution, and the limit of quantification was 6.5 μg mL−1 with a signal-to noise ratio of 10.
3.3 Analysis of samples from various sources
In order to evaluate the characteristic of electropherogram from individual CS origin obtained by EMMA method, we compared the electropherograms of CS originated from cows, pigs and sharks under the optimum conditions (Fig. 6). Each source of CS showed a characteristic electropherogram defined by the content of disaccharides produced by enzyme degradation and these electropherograms were highly reproducible. The obtained disaccharides and their peak percentages are shown in Table 1. Compared with the offline analysis, the established method in this study could detect the low-content disaccharides with a relatively low sample concentration, 500 μg mL−1. The result indicated that CS originated from shark was more sulfated than those from land animals because its abundance in more sulfated disaccharides. The obtained results were in accordance with the previous report.11 In a word, the obtained results revealed that the established EMMA method could be used to identify CS of different origins.
4 Conclusion
An EMMA method was established for the analysis of CS from different sources of cartilage. The degradation of CS in capillary was achieved by employing partial filling mode and a sequence of voltage changes at −1 kV/+1 kV. Results revealed that CS from shark was more sulfated than those from land species, which were in accordance with the previous report. The applicability of the method for the determination of CS concentration was also investigated and a good linearity was obtained. The established method can be used for the characterization of CS samples with minor differences as well as the determination of the CS content.
Acknowledgements
We are grateful for financial support from the “National Natural Science Foundation of China (no. 21205069)”, “the Independent Innovation Fund of Shandong University (no. 2012TS101)” and “the Doctoral Program of Higher Education of Special Research Foundation (The Class of New Teacher) (no. 20110131120039)”.
References
- R. M. Lauder, Compl Ther Med, 2009, 17, 56–62 CrossRef PubMed.
- M. C. Hochberg, N. Engl. J. Med., 2006, 354, 858–860 CrossRef CAS PubMed.
- J. Verges, E. Montell, M. Herrero, C. Perna, J. Cuevas, M. Perez and I. Moller, Dermatol Online J, 2005, 11, 31–33 Search PubMed.
- A. al-Hakim and R. J. Linhardt, Anal. Biochem., 1991, 195, 68–73 CrossRef CAS.
- A. V. Noulas, S. S. Skandalis, E. Feretis, D. A. Theocharis and N. K. Karamanos, Biomed. Chromatogr., 2004, 18, 457–461 CrossRef CAS PubMed.
- F. N. Lamari and N. K. Karamanos, Adv. Pharmacol. (San Diego, CA, U. S.), 2006, 53, 33–48 CAS.
- S. Sakai, E. Otake, T. Toida and Y. Goda, Chem. Pharm. Bull., 2007, 55, 299–303 CrossRef CAS.
- A. Mucci, L. Schenetti and N. Volpi, Carbohydr. Polym., 2000, 41, 37–45 CrossRef CAS.
- W. Garnjanagoonchorn, L. Wongekalak and A. Engkagul, Chem. Eng. Process., 2007, 46, 465–471 CrossRef CAS PubMed.
- H. Zang, L. Li, F. Wang, Q. Yi, Q. Dong, C. Sun and J. Wang, J. Pharm. Biomed. Anal., 2012, 61, 224–229 CrossRef CAS PubMed.
- N. Volpi, Carbohydr. Polym., 2004, 55, 273–281 CrossRef CAS PubMed.
- H. Okamoto, T. Nakajima, Y. Ito, K. Shimada and S. Yamato, J. Chromatogr. A, 2004, 1035, 137–144 CrossRef CAS PubMed.
- C. Bendazzoli, L. Liverani, F. Spelta, M. Prandi, J. Fiori and R. Gotti, J. Pharm. Biomed. Anal., 2010, 53, 1193–1200 CrossRef CAS PubMed.
- C. Bendazzoli, L. Liverani, F. Spelta, M. Prandi, J. Fiori and R. Gotti, J. Pharm. Biomed. Anal., 2010, 53, 1193–1200 CrossRef CAS PubMed.
- C. J. Malavaki, A. P. Asimakopoulou, F. N. Lamari, A. D. Theocharis, G. N. Tzanakakis and N. K. Karamanos, Anal. Biochem., 2008, 374, 213–220 CrossRef CAS PubMed.
- Y. Yang, M. C. Breadmore and W. Thormann, J. Sep. Sci., 2005, 28, 2381–2389 CrossRef CAS PubMed.
- S. Van Dyck, A. Van Schepdael and J. Hoogmartens, Electrophoresis, 2001, 22, 1436–1442 CrossRef CAS.
- B. D. Sanders, R. L. Slotcavage, D. L. Scheerbaum, C. J. Kochansky and T. G. Strein, Anal. Chem., 2005, 77, 2332–2337 CrossRef CAS PubMed.
- L. Pochet, A. C. Servais, E. Farcas, V. Bettonville, C. Bouckaert and M. Fillet, Talanta, 2013, 116, 719–725 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra05576h |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 





