
Unprecedented one-pot multicomponent synthesis of propargylamines using Amberlyst A-21 supported CuI under solvent-free conditions†
Giovanna Bosica* and
John Gabarretta
Department of Chemistry, University of Malta, Msida MSD2080, Malta. E-mail: giovanna.bosica@um.edu.mt
First published on 15th May 2015
Abstract
Amberlyst A-21 supported CuI was found to be a highly efficient novel heterogeneous catalyst for the three-component reaction between amines, aldehydes and alkynes, commonly called A3-coupling. An environmentally benign, one-pot, A3-coupling reaction of various aldehydes, amines and terminal alkynes for the synthesis of propargylamine derivatives is here described. The developed protocol avoids the use of solvent and produces a variety of propargylamines in excellent yields within short reaction times. The catalyst can be easily prepared, recovered and reused for several times, without any appreciable loss in its activity.
Introduction
Propargylamines are highly versatile compounds, serving both as intermediates in the preparation of many nitrogen-containing compounds and also forming key components in a number of bioactive substances, such as polycyclic pyrroles, benzazepines and alkaloids.1–7 Indeed, a number of drugs contain the propargylamine backbone and are mostly targeted at treating neurodegenerative disorders, including the renowned anti-Parkinsonian Rasagiline.6,8,9Traditional routes to propargylamines tended to make use of strong bases such alkyllithium reagents, while other methods feature reactions with isolated imine or enamine intermediates.10–13 However, these methods not only require strict control of reaction conditions and stoichiometry but also occasionally pose challenges in obtaining or handling the required starting materials. By contrast, more recent research has focused on the three-component reaction between amines, aldehydes and alkynes, commonly called A3-coupling (Scheme 1).5,7,14–17 A3-coupling is essentially a combination of the Sonogashira and Mannich reactions, it presents itself as a one-pot multicomponent reaction which facilitates the combination of terminal alkynes with in situ-generated imines.18
 | ||
| Scheme 1 Multicomponent reaction for the formation of propargylamines (4) through A3-coupling – the reaction between amines, aldehydes and alkynes. | ||
One-pot multicomponent coupling reactions (MCRs), where several organic moieties are coupled in one step, are an attractive green synthetic strategy, indeed they are atom economic and efficiently yield the product since the product is formed in one-step instead of multiple sequential steps.19,20 The three-component coupling of aldehydes, amines, and alkynes is an example of MCR and has received much attention in recent years.21 A3-coupling has been demonstrated to occur with a good degree of efficiency under the influence of several salts and complexes of the late transition metals, such as silver, gold, nickel, zinc, iron, cobalt and others,22–27 and different examples of asymmetric variants have also been reported.28–34 Nevertheless, the C–H activation of the terminal alkyne bond has mostly been studied using copper as the catalyst.5,7,14,35 Very often, however, the copper catalysts tend to be employed in homogeneous conditions, with limited opportunity for recovery or re-use of the catalyst. Furthermore, tend to make use of long or harsh reaction conditions, controlled atmospheres or even solvents which are at times environmentally-unfriendly.36–38
This clashes significantly with recent drives towards greener reaction conditions and more efficient catalytic action.39–43 Among greener approaches to A3-coupling reactions particular focus has been made on heterogeneous and recyclable catalysts,44–50 featuring, among others, the use of metal–organic frameworks, clay supports, or in more eccentric cases, eggshell membrane or oyster shell waste.51–55 Indeed, the use of such heterogeneous supports not only enables catalyst immobilisation for easier handling, but also allows for the potential reuse, recycling or regeneration of catalyst surfaces leading to far greener outcomes, as well as more economically viable reactions. Although heterogeneous catalysts studied are quite efficient, there are still some drawbacks which restrict their acceptance, such as high prices of the used precious metals and very often tedious and long preparation methods. Moreover many of the reported heterogeneous catalysed A3-coupling reactions require solvents or microwave assistance.
Therefore, continuing our studies for the development and application of heterogeneous catalysts in new synthetic methods in organic reactions56–58 we set out a clear aim in the identification and use of a financially attractive, recyclable heterogeneous catalyst for the A3-coupling reaction. In order to accomplish the goal of achieving more eco-friendly conditions a set of criteria were set: (a) a cheap, reusable catalyst which is easy to prepare and to handle; (b) no co-catalysts or other auxiliary substances; (c) solvent-free reaction conditions, and (d) high yields and compatibility with a variety of reagents.
Hereunder, the use of a polymer-supported copper catalyst based on copper(I) iodide and Amberlyst A-21 is reported. The catalyst, which, to the best of our knowledge, has not been previously employed in the A3-coupling reaction, demonstrates appreciable substrate flexibility under solvent-free conditions as well as very easy recovery and considerable potential for reusability.
Results and discussion
Catalyst screening and selection
The process of catalyst selection took the form of a series of attempts at the synthesis of product 4a, choosing the coupling reaction of dibutylamine (1a) (1.2 mmol), benzaldehyde (2a) (1 mmol) and phenylacetylene (3a) (1.5 mmol) as model reaction. Table 1 lists the various catalysts employed, all heterogeneous catalysts or catalytic species which could potentially be immobilised onto solid supports. Moreover, species which have been documented to function similarly to the intended A3-coupling reaction were also explored. Nafion NR50, for example has been put forward as a candidate for propargylamine formation, and it was queried whether the SAC-13 form would be viable substitute.59 Alternatively, PdCl2(PPh3)2 has been used extensively in Sonogashira-type cross-coupling reactions which are carried out in copper-free conditions, and it was hoped that it would also be a suitable candidate.60–62 It is evident from the outset, however, that the involvement of a copper-species is quintessential to this reaction proceeding with success. As previously discussed, such copper-based catalysts have been frequently documented in literature as viable promoters of the A3-coupling reaction.5,7,35
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temperature (°C) | Time (h) | Yieldb (%) of 4a |
| a All reactions were carried with dibutylamine (1a) (1.2 mmol), benzaldehyde (2a) (1 mmol) and phenylacetylene (3a) (1.5 mmol).b Refers to yield of isolated pure compound. | |||||
| 1 | PPA–SiO2 (300 wt%) | — | 80 | 24 | — |
| 2 | Nafion SAC-13 (200 wt%) | — | 100 | 24 | — |
| 3 | CeCl3·7H2O (300 wt%) | — | 100 | 24 | — |
| 4 | CuI·A-21 (20 mol%) | — | 100 | 5 | 72 |
| 5 | CuI·A-21 (20 mol%) | Acetonitrile | 100 | 5 | 50 |
| 6 | PdCl2(PPh3)2 (5 mol%) | — | 100 | 2 | — |

The Amberlyst A-21-supported copper(I) iodide indicated in Table 1 has been previously reported to be particularly useful for the Huisgen [3 + 2] cycloaddition for the click synthesis of triazoles.63,64 Given the involvement of alkyne substrates in this reaction, it was postulated that its activity could also be extended to the formation of propargylamines through A3-coupling. These initial outcomes seemed to confirm this hypothesis and reflected the adherence to the main principles and aims set out for the course of this project. As a result, the immobilised copper(I)-based catalyst was selected for the basis of this study. A key observation in this preliminary set of results was the apparent reduction in catalytic activity with the use solvent – in this case acetonitrile – a feature which has been documented in a number of literature sources.65,66 However, this only served to validate the eco-friendly solvent-free conditions in which the reaction was originally intended to be carried out.
Reaction optimisation
On the basis of this encouraging result, efforts were made to optimise the conditions for the reaction. The model reaction selected for this process was the synthesis of 4b (Table 2), a novel product to the best of our knowledge, explaining the shift to 4-methylbenzaldehyde (2b).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Amine (mmol) | Aldehyde (mmol) | Alkyne (mmol) | Catalyst (mol%) | Temperature (°C) | Time (h) | Yieldb (%) of 4b |
| a All reactions carried out in solvent-free conditions under N2 atmosphere using dibutylamine (1a), 4-methylbenzaldehyde (2b) and phenylacetylene (3a).b Refers to yield of isolated pure compound.c Carried out in N2-free conditions for comparison. | |||||||
| 1 | 1.2 | 1 | 1 | 20 | 100 | 5.5 | 70 |
| 2 | 1.2 | 1 | 1 | 10 | 100 | 5.5 | 70 |
| 3 | 1.2 | 1 | 1 | 5 | 100 | 5.5 | 65 |
| 4 | 1.2 | 1 | 1 | 10 | 80 | 5.5 | 67 |
| 5 | 1.2 | 1 | 1 | 10 | 60 | 5.5 | 65 |
| 6 | 1.2 | 1 | 1.2 | 10 | 100 | 5.5 | 77 |
| 7 | 1.2 | 1 | 1.5 | 10 | 100 | 5.5 | 83 |
| 8 | 1.2 | 1 | 1.5 | 20 | 100 | 5.5 | 83 |
| 9 | 1.2 | 1 | 1.5 | 15 | 100 | 5.5 | 79 |
| 10 | 1.2 | 1 | 1.5 | 10 | 100 | 8 | 78 |
| 11 | 1.2 | 1 | 1.5 | 10 | 100 | 18 | 74 |
| 12 | 1.2 | 1 | 1.5 | 10 | R.T. | 168 | — |
| 13 | 1.2 | 1 | 1.5 | 10 | 100 | 5.5 | 82c |

Drastically reducing the amount of catalyst to 5 mol% (entry 3, Table 2), down from 20 mol% (entry 1, Table 2), had a negative effect on the yield of the reaction. A compromise of 10 mol%, however, seemed to be sufficient. Likewise, reduction in the temperature of the reaction also seemed to have a deleterious effect on the yield. In this regard, it was decided that 10 mol% catalyst and a temperature of 100 °C (entry 2, Table 2) should be adopted for the trials that followed.
Tests then shifted to establish the ideal ratio of reagents. Initially, the aim was to have the reaction progress with just an amine excess, for several reasons. Primarily, a reduced number of reagents in excess reflects a lower E-factor, which is more in line with the greener protocol intended here. Secondly, it would also simplify purification of the mixture, given the limited travelling distance in non-polar solvents used for chromatography. Finally, it also signifies a more cost-effective approach. Nevertheless, increasing the proportion of alkyne was seen to have a considerable effect on the final yield of the reaction (entries 6 and 7, Table 2). This could be due to a number of reasons, including to counter evaporation or decomposition at the elevated temperature. There is also the possibility of a side reaction, as shown in Scheme 2, going on which partially consumes the alkyne throughout the reaction. In fact, Glaser-type homocoupling of phenylacetylene is not an uncommon occurrence in the presence of copper(I) catalysts and a base, which in this case could easily have been the amine, and this scenario has also been reported in a similar A3-coupling study by Hell et al., as well as other sources in literature.66,67 For this reason, it was decided that an excess of 0.5 mmol alkyne would be the best approach to ensure that an optimum yield is maintained (entry 7, Table 2).
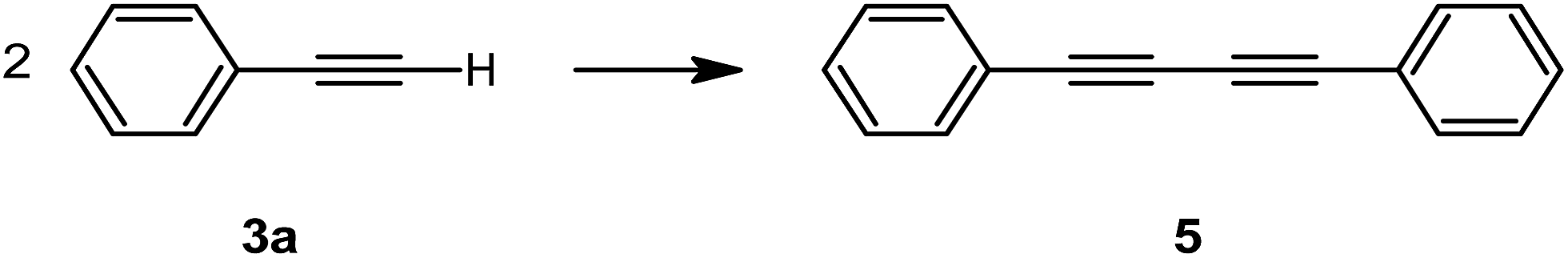 | ||
| Scheme 2 Glaser-type homocoupling as a potential side reaction in A3-coupling justifies an increased excess alkyne to compensate for its consumption. | ||
The tests which followed then focused on confirming that the conditions adopted were indeed the ideal ones. A slight increase in catalyst amount to 15 mol% (entry 9, Table 2) based on this newly-established ratio did not appear to give increased yields, which was treated as a favourable development from a reaction efficiency perspective. Furthermore, increasing the reaction time (entries 10 and 11, Table 2) also appeared not to provide any particular benefits.
When the reaction was attempted at room temperature (entry 12, Table 2), reaction progress was nearly negligible and only a minute portion of the starting materials went into forming the desired product. Using a normal atmosphere (entry 13, Table 2) did not seem to have affected the outcome of the reaction greatly, although it was decided that the nitrogen atmosphere would be retained since some other reagents were air-sensitive.
Being a multicomponent reaction and proceeding successfully through only catalytic amounts of catalyst and with the only by-product being water, the developed protocol displays both a very high Atom Economy as well as a low E-factor, even including water among waste (eqn (1) and (2)). Combined with the lack of solvent and use of a recyclable heterogeneous catalyst, this makes for a very green protocol under these conditions.
 | (1) |
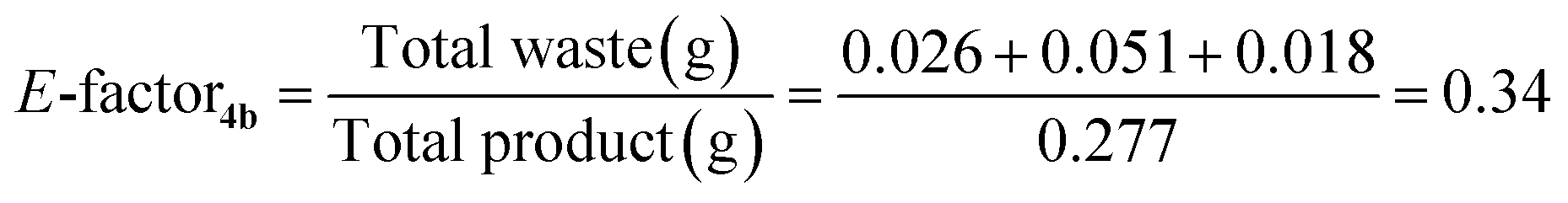 | (2) |
A3-coupling reaction with various amines, aldehydes and alkynes catalysed by CuI·A-21 catalyst
All reactions were performed with 10 mol% of CuI·A-21 catalyst at 100 °C in solvent-free conditions. A series of amines, aldehydes and alkynes were utilised in the synthesis of a small portfolio of propargylamines in line with the general reaction shown in Scheme 1. The initial reaction set (Table 3) involved reacting 4-methylbenzaldehyde (2b) and phenylacetylene (3a) with a variety of cyclic aliphatic amines including piperidine (1b), pyrrolidine (1c) and morpholine (1d) leading to 4c, 4d and 4e in high yields. It is interesting to note the overall improvement on both reaction time and outcome over the use of dibutylamine (1a), a result which most likely boils down to the greater basicity of cyclic amines as well as their relatively compacted structure.5 N-Methylbenzylamine (1e) was employed to explore the effects of aromatic groups within the secondary amine structure, giving 4f in very good yield, while tests with benzylamine (1f) carried out to investigate the reaction's compatibility with primary amines failed to give any discernible results.
|
||||
|---|---|---|---|---|
| Entry | Amine | Time (h) | Product | Yieldb (%) |
| a All reactions carried out in solvent-free conditions under N2 atmosphere using various amines (1b–f) (1.2 mmol), 4-methylbenzaldehyde (2b) (1 mmol) and phenylacetylene (3a) (1.5 mmol).b Refers to yield of isolated pure compound. | ||||
| 1 |  |
3 h |  |
93% |
| 2 |  |
3 h |  |
87% |
| 3 |  |
3 h |  |
96% |
| 4 |  |
3 h | 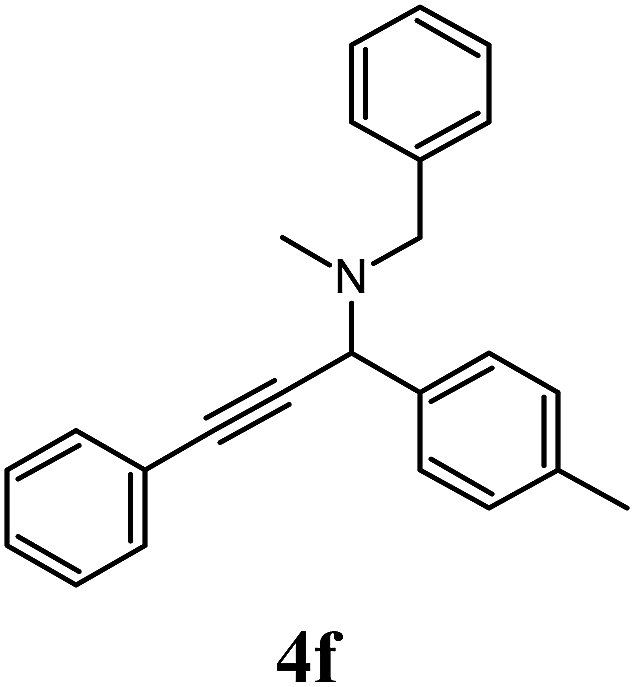 |
95% |
| 5 |  |
6 h |  |
— |

Reactions with aldehydes (Table 4) sought to highlight the flexibility of A3-coupling as a multicomponent reaction. Several aromatic aldehydes, including benzaldehyde (2a) and others with varying para-substituents were used, such as 4-methoxybenzaldehyde (2c) and 4-bromobenzaldehyde (2e), both of which gave very positive results, while furfural (2h) furnished product 4n in an appreciably good yield. In contrast, the reaction with 4-nitrobenzaldehyde (2d) quickly gave a mixture of side-products, turning the mixture black and viscous, something which other sources have also reported.37 High yields were also obtained with the aliphatic aldehydes valeraldehyde (2f) and heptaldehyde (2g).
|
||||
|---|---|---|---|---|
| Entry | Aldehyde | Time (h) | Product | Yieldb (%) |
| a All reactions carried out in solvent-free conditions under N2 atmosphere using piperidine (1b) (1.2 mmol), various aldehydes (2a, c–h) (1 mmol) and phenylacetylene (3a) (1.5 mmol).b Refers to yield of isolated pure compound. | ||||
| 1 |  |
3 h |  |
95% |
| 2 |  |
2 h |  |
98% |
| 3 |  |
3 h |  |
— |
| 4 |  |
2 h |  |
94% |
| 5 |  |
2 h |  |
88% |
| 6 |  |
2 h |  |
88% |
| 7 |  |
2 h | 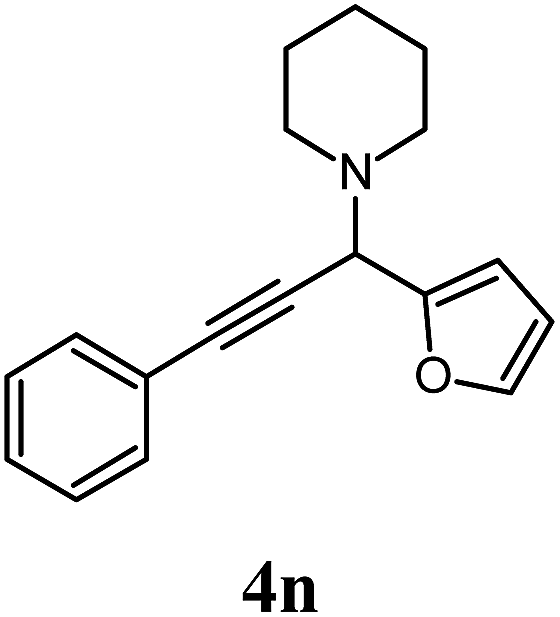 |
82% |

The alkyne set (Table 5) sought to briefly demonstrate reaction viability with different kinds of alkynes. The reaction shows compatibility with both alkyl and substituted aryl alkynes as evidenced by the appreciable yields with 1-octyne (3b) and 4-methylphenylacetylene (3c). A3-coupling was also effected with the silyl alkyne (triisopropylsilyl)acetylene (3d). The lower yield in this case could potentially be attributed to the competing Glaser homocoupling reaction, as well as a slower reaction due to the relative steric effects of the bulky isopropyl groups.






A final set of A3-coupling reactions (Table 6) focused on demonstrating the opportunities for varied synthesis, characteristic of such multicomponent reactions. Selected substrates were employed to give a variety of propargylamine products and helped to shed more light on the nature of the A3-coupling reaction. The synthesis of 4r, for example, afforded the product in high yield within a very short timeframe when compared to its benzaldehyde (2a) (Table 1) and 4-methylbenzaldehyde (2c) (Table 2) analogues. On the other hand, a longer reaction time and lower yield was observed when using 1-hexyne (3e) as the alkyne substituent. Nevertheless, as with the formation of product 4f, the reaction with N-methylbenzylamine (1e) gave product 4t in high yield.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Amine | Aldehyde | Alkyne | Time (h) | Product | Yieldb (%) |
| a All reactions carried out in solvent-free conditions under N2 atmosphere using various amines (1.2 mmol), aldehydes (1 mmol) and alkynes (1.5 mmol).b Refers to yield of isolated pure compound.c Butyraldehyde (2i) used as aldehyde substrate.d 1-Hexyne (3e) used as alkyne substrate. | ||||||
| 1 | 1a | 2ic | 3a | 2 h |  |
92% |
| 2 | 1a | 2b | 3ed | 6 h |  |
80% |
| 3 | 1e | 2g | 3a | 4 h |  |
93% |

Some mechanistic aspects
Several tentative mechanisms for propargylamine formation through A3-coupling have been put forward. Most, however, tend to focus on the involvement of copper acetylides (7) (Fig. 1) which go on to react with iminium ions (6) formed in situ between the aldehyde and secondary amines (1).35,37,68 In reality, the proposed acetylide formation step may also involve the formation of a π-metal–alkyne complex intermediate, a step which is considered critical in increasing terminal proton's acidity to facilitate its abstraction. Incidentally this process greatly resembles the initial steps in Sonogashira-type processes, and copper-based catalysts, or co-catalysts, are in fact traditional key-players in these type of reactions, as exemplified in a recent study by Zou et al., for instance.69 | ||
| Fig. 1 Proposed reaction mechanism for the formation of propargylamines (4) by means of CuI·A-21 catalyst. | ||
The nature of the catalyst itself provided an interesting insight into this mechanistic detail since an immediate colour change of the catalyst, from greenish to bright yellow, was observed as soon as the flask containing the reagents mixture was immersed into the warm oil bath. Upon closer inspection in further tests, this was noted to start as soon as the alkyne was added to the mixture and progressed even before direct heating was applied (Fig. 2). In fact, literature sources tend to highlight the formation of this bright yellow colour under these conditions as evidence of copper acetylides.70 Similar observations have also been noted by Albaladejo's group working on A3-coupling with copper nanoparticles immobilised on titanium(IV) oxide.65
 | ||
| Fig. 2 Images of CuI·A-21 before [left] and after [right] addition of alkyne to reaction mixture. Colour change from green to yellow suggests copper acetylide formation. | ||
Catalyst activity, stability and recyclability
The preparation of the CuI·A-21 catalyst resulted in a clear colour change from white to greenish (Fig. 3). This was assumed to be a result of successful fixation of the copper salt onto the polymer, most likely by chelation (Scheme 3).63,64 | ||
| Fig. 3 Images of Amberlyst A-21 resin beads before [left] and after [right] addition of copper(I) iodide. | ||
 | ||
| Scheme 3 Reaction scheme for the formation of copper(I) iodide on Amberlyst A-21. | ||
Immobilisation of the active copper species brought with it numerous benefits (Fig. 4). The main advantage of the Amberlyst-based catalyst was the beaded nature of the resin. This made the catalyst very easy to handle, especially during filtration and recovery steps, and even if some of the catalyst was accidentally dropped onto the bench, recovery was greatly facilitated. Furthermore, the polymer support itself improves the green protocol of the reaction, as evidenced by data from a test carried out with unsupported copper(I) iodide (Table 7). Although the starting quantities and GC yields emerged to be identical, the advantage of the polymer support is evident, and gives an E-factor of just 0.34. Making use of the unsupported catalyst, on the other hand, gives a higher E-factor and also results in the loss of the catalyst, which diminishes the cost-effectiveness of the procedure.
 | ||
| Fig. 4 Key advantages of CuI·A-21 as a heterogeneous catalyst for the A3-coupling reaction. | ||
| Unsupported CuI | CuI·A-21 | |
|---|---|---|
| a Based on synthesis of product 4b from 1a, 2b and 3a as in Table 2.b GC Yield. | ||
| Reagent ratioa | 1 (Aldehyde)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 (amine):1.5 (alkyne) :1.2 (amine):1.5 (alkyne) |
|
| Catalyst quantity | 20 mol% | |
| Yieldb | 85% | |
| Workup | Direct column loading | Catalyst filtration |
| Waste | 0.2 mmol CuI catalyst | |
| 0.2 mmol amine excess | 0.2 mmol amine excess | |
| 0.5 mmol alkyne excess | 0.5 mmol alkyne excess | |
| 1 mmol H2O | 1 mmol H2O | |
| E-factor | 0.47 including loss of catalyst | 0.34 with complete catalyst recovery |
Apart from this, the nature of the catalyst also offers the added advantage of homogeneous immobilisation of nano-sized copper which maximises surface area within the macroporous polymer network. The fact that copper is most likely chelated by the terminal nitrogen atoms within the polymer network is also known to prevent oxidation or disproportionation of the metal ion, while improving its catalytic activity.64
The catalyst itself displayed high stability and could easily be handled in air. In fact, batches of CuI·A-21 which were left on shelves for the duration of their use – approximately three months – remained equally active without any discernible physical changes. The nitrogenous atmosphere employed during the trials was used exclusively to ensure that any air-sensitive reagents did not decompose, and did not seem to have any influence on the catalyst's performance.
An additional advantage of the Amberlyst A-21 support is that it offers the possibility of recharging the catalyst with more copper(I) iodide should this be required, meaning the lifetime of its recyclability can be extended.64
Catalyst recycling trials (Fig. 5) also indicated it was able to retain a largely consistent performance over around five consequent reaction cycles, before its activity began to drop considerably.
 | ||
| Fig. 5 Recycling trials of CuI·A-21 catalyst in the synthesis of product 4h show consistent performance over 5 cycles. | ||
Experimental
General
All reagents are commercially available (Aldrich, Acros, Alfa Aesar, BDH, Fischer Scientific) and were used without further purification. Silica on PET sheets containing a fluorescent indicator, fluorescing at 254 nm under UV irradiation, was used as a stationary phase for TLC. Mixtures of n-hexane and ethyl acetate were as the mobile phase. The TLC sheets were viewed under UV light at a wavelength of 254 nm, followed by treatment with a basic potassium permanganate solution.Gas chromatography (GC) was performed using a Shimadzu GC-2010 plus gas chromatograph using a flame ionisation detector (FID) and nitrogen as the carrier gas. Samples were dissolved in a small amount of solvent prior to being injected manually. The capillary column, a HiCap 5 (5% phenyl–95% methylpolysiloxane) column having dimensions of 0.32 mm (internal diameter) × 30 m (length) × 0.25 μm (film thickness), was used with a temperature program of 65–300 °C at a heating rate of 15 °C min−1. Gas chromatography-mass spectrometry (GC-MS) was carried out by means of a Thermo Finnigan Trace DSQ quadropole mass spectrometer together with a Thermo Finnigan Trace GC Ultra equipped with a 25 m × 0.22 mm BP1 (100% dimethylpolysiloxane stationary phase) column. The temperature program used for this set of analyses was identical to the utilised GC parameters. Gas chromatography-mass spectrometry (GC-MS) of products 4o, 4q and 4r was carried out by means of a Hewlett-Packard 5973 series mass selective detector together with a Agilent 6890 series GC equipped with a Hewlett-Packard 7683 series auto-sampler and a 30 m × 0.32 mm × 1 μm HP1 (100% dimethylpolysiloxane stationary phase) column. The temperature program used for this set of analyses was of 100–290 °C at a heating rate of 10 °C min−1. NMR analysis was performed using a Bruker AM250 NMR spectrometer fitted with a dual probe at frequencies of 250 MHz for 1H NMR and 62.9 MHz for 13C NMR. Processing was carried out using an Aspect 3000 computer having 16 K and 64 K complex points for 1H and 13C NMR respectively. Deuterated chloroform with TMS internal standard was used to dissolve samples as follows: 5 mg in 0.8 mL CDCl3 for 1H samples and around 35–40 mg in 0.8 mL CDCl3 for 13C samples. IR spectroscopy was carried out using a Shimadzu IRAffinity-1 FTIR spectrometer, calibrated against a 1602 cm−1 polystyrene absorbance spectrum. Samples were prepared as thin films in between sodium chloride discs. Microanalyses were performed with a CHNS–O analyzer Model EA 1108 from Fisons Instruments. Melting point determination of solid products (4e and 4p) was carried out using a Gallenkamp melting point determination apparatus fitted with a mercury thermometer. Three consecutive melting point tests were carried out to ensure consistency. Many of the synthesised compounds are known and their spectroscopic data are in agreement with those reported in the literature.
General procedure for A3-coupling reactions
The general procedure for the A3-coupling reactions was based on the optimised conditions as selected in the synthesis of product 4b. A nitrogen-flushed 25 mL two-necked flask was loaded with 10 mol% of CuI·A-21 catalyst, to which were then added the respective amine (1) (1.2 mmol), aldehyde (2) (1 mmol) and alkyne (3) (1.5 mmol) in that order. After a few minutes of mixing at room temperature, the mixture was then stirred at 100 °C using an oil bath and reflux condenser in the absence of solvent. Reactions were monitored by TLC and GC. Once the aldehyde was seen to have disappeared completely, or no further change in the progress of the reaction was noted, the reaction was stopped by cooling to room temperature and the catalyst filtered off using two 5 mL portions of diethyl ether. The catalyst was then washed with a further two 5 mL portions of diethyl ether. The resulting products (4) were then purified by column chromatography, using mixtures of n-hexane and ethyl acetate, and dried under vacuum. Yields were obtained and the products were analysed by 1H and 13C NMR spectroscopy, IR spectroscopy as well as gas chromatography-mass spectrometry and elemental analyses where available.Preparation of copper(I) iodide on Amberlyst A-21 (CuI·A-21)
A sample of dry Amberlyst A-21 was prepared by placing 5 g of the resin in 25 mL methanol and allowing it to stand for 30 minutes. After this, the mixture was filtered and washed with 10 mL methanol three times over. This procedure was then repeated in dichloromethane, and the resin was then placed in a vacuum desiccator to dry overnight.A solution of 381 mg (2 mmol) of copper(I) iodide in 15 mL acetonitrile was then mixed with 1 g of dry Amberlyst A-21 and left stirring overnight at room temperature. The solvent was then evaporated off and the light green resin was washed with two 15 mL aliquots of acetonitrile, followed by two 15 mL aliquots of dichloromethane. The resin was then dried overnight in a vacuum desiccator and, after confirming a stable weight, the loading of copper per gram of resin calculated by observing the weight increase of the final dried sample of CuI·A-21.
Analytical data
:1, 72%, 230 mg). IR (neat, cm−1) ν: 3061 (m), 3030 (m), 2955 (s), 2930 (s), 2860 (s), 2818 (s), 1948 (w), 1599 (m), 1489 (s), 1466 (s), 1448 (s), 1377 (m), 1323 (m), 1298 (m), 1273 (m), 1175 (m), 1155 (m), 1096 (m), 1068 (m), 1028 (m), 962 (m), 912 (m), 754 (s), 723 (s), 690 (s). 1H NMR (250 MHz, CDCl3): δ 7.68–7.27 (m, 10H), 5.02 (s, 1H), 2.49 (t, J = 7.3 Hz, 4H), 1.56–1.16 (m, 8H), 0.84 (t, J = 7.3 Hz, 6H). 13C NMR (62.9 MHz, CDCl3): δ 140.1, 132.0, 128.6, 128.4, 128.2, 128.1, 127.3, 123.7, 87.9, 86.3, 57.8, 50.8, 30.5, 20.7, 14.2.:1, 83%, 277 mg). IR (neat, cm−1) ν: 3053 (m), 3022 (m), 2955 (s), 2928 (s), 2870 (m), 2860 (m), 2818 (m), 1597 (m), 1508 (m), 1489 (m), 1456 (m), 1443 (m), 1377 (m), 1317 (m), 1316 (m), 1296 (m), 1271 (m), 1176 (m), 1155 (m), 1096 (m), 1068 (m), 1022 (m), 968 (w), 943 (w), 912 (w), 754 (s), 690 (s). 1H NMR (250 MHz, CDCl3): δ, 7.56–7.14 (m, 9H), 4.98 (s, 1H), 2.49 (t, J = 7.3 Hz, 4H), 2.35 (s, 1H), 1.55–1.09 (m, 8H), 0.85 (t, J = 7.3 Hz, 6H). 13C NMR (62.9 MHz, CDCl3): δ 137.8, 137.5, 132.6, 129.4, 129.1, 128.7, 12.4, 88.1, 87.4, 58.1, 51.4, 31.3, 21.9, 21.3, 14.8. GC-MS (r.t. 18.58 min): m/z (%) = 333 (1) [M]+, 290 (16), 276 (1), 242 (100), 219 (1), 205 (57), 191 (16), 165 (10), 130 (13), 114 (26), 91 (20), 77 (8), 57 (11), 41 (17). Anal. calcd for C24H31N (333.5097): C, 86.4; H, 9.4. Found: C, 86.3; H, 9.5.:1, 93%, 268 mg). IR (neat, cm−1) ν: 3053 (m), 3022 (m), 2932 (s), 2853 (m), 2804 (m), 1684 (m), 1597 (m), 1508 (s), 1489 (s), 1465 (m), 1443 (s), 1319 (s), 1292 (m), 1271 (s), 1203 (m), 1175 (m), 1153 (m), 1113 (m), 1092 (s), 1068 (m), 989 (m), 966 (m), 847 (m), 824 (m), 756 (s), 690 (s). 1H NMR (250 MHz, CDCl3): δ = 7.81–7.69 (m, 9H), 4.75 (s, 1H), 2.63–2.46 (m, 4H), 2.35 (s, 3H), 1.65–1.51 (m, 4H), 1.59–1.45 (m, 2H); 13C NMR (62.9 MHz, CDCl3): δ = 137.1, 135.7, 131.8, 128.8, 128.5, 128.3, 128.0, 123.5, 87.6, 86.4, 62.2, 50.7, 26.2, 24.5, 21.1.:3, 87%, 240 mg). IR (neat, cm−1) ν: 3053 (m), 3022 (m), 2934 (s), 2929 (m), 2874 (m), 1684 (m), 1599 (m), 1510 (s), 1489 (s) 1456 (m), 1443 (m), 1294 (m), 1271 (m), 1269 (m), 1175 (m), 1128 (m), 1111 (m), 1022 (m), 974 (w), 822 (m), 756 (s), 690 (s). 1H NMR (250 MHz, CDCl3): δ = 7.54–7.11 (m, 9H), 4.86 (s, 1H), 2.75–2.63 (m, 4H), 2.35 (s, 3H), 1.85–1.73 (m, 4H); 13C NMR (62.9 MHz, CDCl3): δ = 137.2, 136.4, 131.8, 129.0, 128.3, 128.0, 123.5, 86.9, 86.8, 58.9, 50.3, 23.5, 21.1.:15, 96%, 280 mg). mp 79–80 °C. IR (neat, cm−1) ν: 3053 (w), 3022 (w), 2954 (m), 2922 (m), 2912 (m), 2893 (m), 2853 (m), 2820 (m), 1508 (m), 1489 (m) 1456 (m), 1443 (m), 1319 (m), 1286 (m), 1273 (m), 1246 (m), 1117 (s), 1070 (m), 1022 (w), 1005 (m), 970 (w), 930 (m), 866 (m), 849 (m), 825 (w), 756 (s), 690 (s). 1H NMR (250 MHz, CDCl3): δ = 7.56–7.13 (m, 9H), 4.75 (s, 1H), 3.81–3.64 (m, 4H), 2.72–2.54 (m, 4H), 2.36 (s, 3H); 13C NMR (62.9 MHz, CDCl3): δ = 137.5, 134.9, 131.8, 128.9, 128.5, 128.3, 128.2, 123.1, 88.3, 85.3, 67.2, 61.8, 49.9, 21.1.:15, 95%, 308 mg). IR (neat, cm−1) ν: 3082 (m), 3059 (m), 3028 (m), 3001 (m), 2978 (m), 2943 (m), 2922 (m), 2840 (m), 2791 (m), 1948 (w), 1801 (w), 1597 (m), 1510 (m), 1489 (s), 1454 (s), 1443 (m), 1416 (m), 1363 (m), 1325 (m), 1319 (m), 1294 (m), 1274 (m), 1246 (w), 1209 (w), 1188 (m), 1175 (m), 1122 (m), 1070 (m), 1018 (s), 970 (m), 912 (m), 852 (m), 802 (m), 771 (m), 756 (s), 741 (m), 690 (s). 1H NMR (250 MHz, CDCl3): δ = 7.62–7.12 (m, 14H), 4.89 (s, 1H), 3.62, 3.71 (ABq, JAB = 13.1 Hz, 2H), 2.35 (s, 3H), 2.24 (s, 3H); 13C NMR (62.9 MHz, CDCl3): δ = 139.4, 137.1, 136.1, 131.9, 129.0, 128.8, 128.3, 128.3, 128.1, 127.0, 123.4, 88.4, 85.1, 59.4, 58.8, 38.0, 21.1. GC-MS (r.t. 19.92 min): m/z (%) = 325 (8) [M]+, 310 (1), 234 (28), 205 (100), 189 (13), 165 (7), 118 (7), 91 (26), 77 (6), 65 (8). Anal. calcd for C24H23N (325.4462): C, 88.6; H, 7.1. Found: C, 88.4; H, 7.2.:15, 95%, 260 mg). IR (neat, cm−1) ν: 3061 (m), 3030 (m), 2931 (s), 2852 (s), 2804 (s), 2748 (m), 1948 (w), 1597 (m), 1570 (w), 1489 (s), 1466 (m), 1443 (s), 1321 (m), 1290 (m), 1271 (m), 1201 (m), 1153 (m), 1096 (m), 1068 (m), 1028 (m), 991 (m), 968 (m), 914 (m), 864 (m), 789 (m), 754 (s), 690 (s). 1H NMR (250 MHz, CDCl3): δ = 7.67–7.28 (m, 9H), 4.80 (s, 1H), 2.64–2.47 (m, 4H), 1.71–1.51 (m, 4H), 1.50–1.37 (m, 2H); 13C NMR (62.9 MHz, CDCl3): δ = 138.7, 132.5, 131.8, 128.5, 128.3, 128.0, 127.4, 123.4, 87.8, 86.1, 62.4, 50.7, 26.2, 24.5.:15, 98%, 299 mg). IR (neat, cm−1) ν: 3061 (m), 3034 (m), 2997 (m), 2932 (s), 2853 (s), 2833 (s), 2804 (s), 2748 (m), 1890 (w), 1737 (m), 1688 (m), 1610 (s), 1598 (s), 1583 (s), 1504 (s), 1489 (s), 1454 (s), 1443 (s), 1319 (s), 1302 (s), 1247 (s), 1169 (s), 1113 (s), 1092 (s), 1068 (m), 1038 (s), 989 (m), 968 (m), 914 (m), 848 (m), 773 (s), 756 (s), 690 (s). 1H NMR (250 MHz, CDCl3): δ = 7.57–7.27 (m, 7H), 6.94–6.84 (m, 2H), 4.75 (s, 1H), 3.83 (s, 3H), 2.62–2.45 (m, 4H), 1.71–1.50 (m, 4H), 1.49–1.37 (m, 2H); 13C NMR (62.9 MHz, CDCl3): δ = 159.0, 131.8, 130.8, 129.6, 128.3, 128.0, 123.5, 113.4, 87.6, 86.5, 61.8, 55.3, 50.6, 26.2, 24.5.:5, 94%, 332 mg). IR (neat, cm−1) ν: 3053 (m), 2934 (s), 2853 (s), 2833 (s), 2806 (s), 2748 (m), 1902 (w), 1737 (w), 1597 (s), 1583 (m), 1504 (s), 1574 (m), 1483 (s), 1466 (m), 1443 (s), 1396 (s), 1317 (m), 1286 (m), 1269 (m), 1202 (m), 1171 (w), 1153 (m), 1113 (m), 1092 (s), 1070 (s), 1013 (s), 991 (s), 970 (m), 912 (w), 848 (m), 806 (m), 754 (s), 690 (s). 1H NMR (250 MHz, CDCl3): δ = 7.59–7.27 (m, 9H), 4.74 (s, 1H), 2.62–2.45 (m, 4H), 1.71–1.50 (m, 4H), 1.49–1.37 (m, 2H); 13C NMR (62.9 MHz, CDCl3): δ = 137.9, 131.8, 131.2, 130.2, 128.3, 128.2, 123.1, 121.4, 88.3, 85.3, 61.8, 50.7, 26.2, 24.4.:1, 88%, 225 mg). IR (neat, cm−1) ν: 3055 (w), 2932 (s), 2856 (s), 2803 (s), 2748 (w), 2681 (w), 1942 (w), 1597 (m), 1588 (s), 1489 (m), 1466 (m), 1452 (m), 1443 (s), 1325 (m), 1304 (w), 1256 (m) 1155 (m), 1116 (m), 1096 (m), 1068 (w), 910 (w), 862 (w), 754 (s), 690 (s). 1H NMR (250 MHz, CDCl3): δ = 7.49–7.26 (m, 5H), 3.52–3.42 (m, 1H), 2.72–2.60 (m, 2H), 2.52–2.40 (m, 2H), 1.84–1.19 (m, 12H), 0.93 (t, J = 7.3 Hz, 3H); 13C NMR (62.9 MHz, CDCl3): δ = 131.8, 128.2, 127.7, 123.7, 88.3, 85.6, 58.7, 50.6, 33.5, 29.1, 26.2, 24.6, 22.6, 14.1.:1, 88%, 250 mg). IR (neat, cm−1) ν: 3055 (w), 2932 (s), 2856 (s), 2803 (s), 2748 (w), 2681 (w), 1942 (w), 1597 (m), 1588 (s), 1489 (m), 1466 (m), 1452 (m), 1443 (s), 1325 (m), 1304 (w), 1155 (m), 1116 (m), 1096 (m), 1068 (w), 910 (w), 862 (w), 754 (s), 690 (s). 1H NMR (250 MHz, CDCl3): δ = 7.49–7.26 (m, 5H), 3.52–3.42 (m, 1H), 2.76–2.60 (m, 2H), 2.57–2.40 (m, 2H), 1.84–1.19 (m, 16H), 0.89 (t, J = 6.7 Hz, 3H); 13C NMR (62.9 MHz, CDCl3): δ = 131.8, 128.2, 127.7, 123.7, 88.3, 85.6, 58.7, 50.6, 33.5, 31.8, 29.1, 26.9, 26.2, 24.6, 22.6, 14.1.:15, 82%, 216 mg). IR (neat, cm−1) ν: 3115 (w), 3055 (w), 2934 (m), 2806 (s), 2852 (s), 2748 (m), 1599 (m), 1558 (s), 1489 (s), 1468 (m), 1452 (m), 1443 (s), 1317 (m), 1300 (m), 1225 (m), 1206 (m), 1184 (m), 1153 (m), 1142 (m), 1115 (m), 1092 (m), 1070 (w), 1012 (s), 939 (m), 814 (m), 885 (m), 777 (m), 754 (s), 735 (s), 690 (s). 1H NMR (250 MHz, CDCl3): δ = 7.54–7.46 (m, 2H), 7.45–7.41 (m, 1H), 7.38–7.29 (m, 3H), 6.51–6.46 (m, 1H), 6.38–6.33 (m, 1H), 4.88 (s, 1H), 2.68–2.50 (m, 4H), 1.76–1.52 (m, 4H), 1.51–1.36 (m, 2H); 13C NMR (62.9 MHz, CDCl3): δ = 151.8, 142.6, 131.9, 128.3, 128.3, 123.0, 110.0, 109.2, 86.4, 83.9, 56.6, 50.6, 26.0, 24.4.:1, 78%, 231 mg). IR (neat, cm−1) ν: 3051 (m), 3024 (m), 2932 (s), 2855 (s), 2804 (s), 2747 (m), 2257 (w), 1903 (w), 1707 (m), 1604 (w), 1510 (s), 1466 (s), 1454 (s), 1443 (s), 1379 (m), 1319 (s), 1296 (s), 1269 (s), 1204 (m), 1175 (m), 1155 (s), 1113 (s), 1088 (s), 1064 (m), 1038 (m), 1022 (m), 989 (s), 848 (m), 823 (m), 808 (m), 764 (s), 725 (m), 658 (m). 1H NMR (250 MHz, CDCl3): δ = 7.42 (d, J = 7.6 Hz, 2H), 7.13 (d, J = 7.6 Hz, 2H), 4.49 (s, 1H), 2.52–2.36 (m, 4H), 2.34 (s, 3H), 2.31–2.24 (m, 4H), 1.66–1.23 (m, 12H), 0.90 (t, J = 6.7 Hz, 3H); 13C NMR (62.9 MHz, CDCl3): δ = 136.8, 136.3, 128.6, 128.5, 87.7, 61.8, 50.5, 31.4, 29.1, 28.6, 26.2, 24.5, 22.6, 21.1, 18.8, 14.0. GC-MS (r.t. 19.25 min): m/z (%) = 297 (18) [M]+, 282 (1), 226 (8), 212 (10), 206 (100), 128 (10), 142 (10), 105 (20), 84 (2). Anal. calcd for C21H31N (297.4776): C, 84.8; H, 10.5. Found: C, 84.6; H, 10.7.:1, 94%, 285 mg). mp 73–74 °C. IR (neat, cm−1) ν: 3051 (m), 3026 (w), 2932 (m), 2856 (m), 2804 (w), 2747 (w), 1734 (m), 1717 (w), 1684 (w), 1506 (s), 1456 (m), 1454 (s), 1443 (s), 1379 (m), 1319 (w), 1271 (m), 1115 (m), 1092 (m), 815 (s), 764 (m). 1H NMR (250 MHz, CDCl3): δ = 7.50 (d, J = 7.9 Hz, 2H), 7.40 (d, J = 8.6 Hz, 2H), 7.15 (t, J = 7.9 Hz, 4H), 4.74 (s, 1H), 2.62–2.46 (m, 4H), 2.36 (s, 6H), 2.31–2.24 (m, 4H), 1.66–1.50 (m, 4H), 1.49–1.35 (m, 2H); 13C NMR (62.9 MHz, CDCl3): δ = 138.0, 137.0, 135.8, 131.7, 130.0, 128.7, 128.5, 120.4, 87.6, 85.7, 62.2, 50.7, 26.3, 24.5, 21.4, 21.1. GC-MS (r.t. 18.93 min): m/z (%) = 303 (22) [M]+, 288 (2), 219 (100), 212 (28), 203 (14), 189 (8), 165 (4), 129 (6), 91 (1), 65 (2), 41 (2). Anal. calcd for C22H25N (303.4406): C, 87.1; H, 8.3. Found: C, 87.3; H, 8.6.:1, 70%, 257 mg). IR (neat, cm−1) ν: 2940 (s), 2891 (s), 2864 (s), 2806 (m), 2747 (m), 2158 (m), 1558 (m), 1510 (m), 1464 (s), 1443 (m), 1383 (m), 1364 (m), 1317 (m), 1294 (m), 1267 (m), 1202 (m), 1175 (m), 1153 (m), 1113 (m), 1092 (m), 1003 (s), 988 (s), 920 (m), 883 (s), 847 (m), 822 (m), 808 (), 766 (s), 721 (s), 677 (s). 1H NMR (250 MHz, CDCl3): δ = 7.49 (d, J = 7.9 Hz, 2H), 7.14 (d, J = 7.9 Hz, 2H), 4.63 (s, 1H), 2.55–2.40 (m, 4H), 2.34 (s, 3H), 1.65–1.48 (m, 4H), 1.47–1.33 (m, 2H), 1.19–1.02 (m, 21H); 13C NMR (62.9 MHz, CDCl3): δ = 136.9, 135.8, 128.6, 128.4, 103.8, 87.9, 62.3, 50.3, 26.3, 24.6, 21.1, 18.8, 11.4. GC-MS (r.t. 20.46 min): m/z (%) = 369 (10) [M]+, 326 (2), 278 (67), 212 (16), 207 (100), 191 (12), 159 (16), 157 (6), 133 (10), 111 (10), 96 (10), 84 (8), 73 (12). Anal. calcd for C24H39NSi (369.6587): C, 79.0; H, 10.6. Found: C, 78.7; H, 10.4.:5, 92%, 262 mg). IR (neat, cm−1) ν: 3055 (w), 2957 (s), 2930 (s), 2872 (s), 2816 (m), 1944 (w), 1489 (s), 1466 (s), 1456 (m), 1443 (m), 1377 (m), 1310 (m), 1273 (m), 1250 (m), 1173 (m), 1113 (m), 1090 (m), 1070 (m), 1028 (m), 910 (m), 883 (s), 756 (s), 690 (s). 1H NMR (250 MHz, CDCl3): δ = 7.46–7.20 (m, 5H), 3.64 (t, J = 7.3 Hz, 1H), 2.65–2.48 (m, 2H), 2.47–2.32 (m, 2H), 1.72–1.18 (m, 6H), 0.95 (t, J = 7.3 Hz, 3H), 0.92 (t, J = 6.7 Hz, 6H); 13C NMR (62.9 MHz, CDCl3): δ = 131.7, 128.2, 127.6, 123.9, 89.4, 84.4, 53.9, 51.4, 36.5, 30.9, 20.7, 20.0, 14.1, 13.9. GC-MS (r.t. 15.52 min): m/z (%) = 285 (1) [M]+, 270 (1), 242 (100), 213 (1), 186 (12), 158 (13), 141 (5), 129 (19), 114 (61), 91 (11), 77 (7), 65 (3), 57 (9), 41 (17). Anal. calcd for C20H31N (285.4669): C, 84.15; H, 10.95. Found: C, 84.3; H, 10.8.:5, 80%, 251 mg). IR (neat, cm−1) ν: 3022 (w), 2957 (s), 2930 (s), 2860 (s), 2818 (s), 2257 (w), 1904 (w), 1510 (s), 1458 (s), 1377 (m), 1319 (m), 1296 (m), 1267 (m), 1176 (m), 1096 (m), 1066 (m), 1022 (m), 943 (m), 823 (m), 772 (m), 737 (s). 1H NMR (250 MHz, CDCl3): δ = 7.48 (d, J = 7.9 Hz, 2H), 7.12 (d, J = 7.9 Hz, 2H), 4.73 (s, 1H), 2.44–2.21 (m, 6H), 2.34 (s, 3H), 1.64–1.11 (m, 12H), 0.94 (t, J = 7.3 Hz, 3H), 0.83 (t, J = 6.7 Hz, 6H); 13C NMR (62.9 MHz, CDCl3): δ = 137.7, 136.4, 128.4, 128.3, 87.2, 79.3, 56.7, 50.5, 31.3, 30.5, 22.0, 21.1, 20.5, 18.5, 14.0, 13.6. GC-MS (r.t. 19.25 min): m/z (%) = 313 (4) [M]+, 270 (24), 256 (4), 232 (1), 222 (4), 185 (100), 156 (4), 141 (10), 128 (19), 57 (6). Anal. calcd for C22H35N (313.52): C, 84.3; H, 11.25. Found: C, 84.5; H, 11.4.:5, 93%, 296 mg). IR (neat, cm−1) ν: 3062 (m), 3030 (m), 2953 (s), 2930 (s), 2856 (s), 2793 (m), 1597 (m), 1489 (s), 1454 (m), 1443 (m), 1364 (w), 1340 (w), 1321 (w), 1252 (w), 1209 (w), 1144 (w), 1124 (w), 1096 (w), 1070 (m), 1028 (m), 968 (w), 912 (w), 827 (w), 756 (s), 739 (m), 690 (s). 1H NMR (250 MHz, CDCl3): δ = 7.51–7.20 (m, 10H), 3.55 (t, J = 7.3 Hz, 1H), 3.73, 3.54 (ABq, JAB = 13.1 Hz, 2H), 2.29 (s, 3H), 1.81–1.67 (m, 2H), 1.61–1.40 (m, 2H), 1.38–1.20 (m, 6H), 0.88 (t, J = 6.7 Hz, 3H); 13C NMR (62.9 MHz, CDCl3): δ = 139.5, 131.8, 129.0, 128.3, 128.2, 127.8, 126.9, 123.6, 123.4, 87.7, 85.7, 59.3, 56.0, 37.8, 34.0, 31.8, 29.0, 26.5, 22.6, 14.1.Conclusions
In summary, it was found that CuI·A-21 was a suitable novel catalyst for the one-pot synthesis of propargylamines from aldehydes, amines and alkynes through A3-coupling. This was carried out in solvent-free conditions to yield an assortment of propargylamines in good to excellent yields (70–98%). The catalyst proved to be very stable and could easily be prepared from relatively cheap starting materials and subsequently stored for considerable periods of time. Recycling tests indicate that the catalyst can be used for five consecutive cycles without displaying a significant drop in activity. Under the developed conditions, the reaction displays a very ‘green’ protocol with high Atom Economy of roughly 95% and a low E-factor of around 0.3 in most cases and by totally excluding the need for toxic or dangerous solvents while also having the added advantage of a recyclable heterogeneous catalyst, containing neither noble nor heavy or toxic metals, and relatively short reaction times, overall opening true opportunities for practical application.Acknowledgements
The Authors thank the University of Malta and the Strategic Educational Pathways Scholarship – Malta (European Union – European Social Fund (ESF) under Operational Programme II – Cohesion Policy 2007–2013, “Empowering People for More Jobs and a Better Quality Of Life”) for financial support.References
- G. S. Kauffman, G. D. Harris, R. L. Dorow, B. R. P. Stone, R. L. Parsons Jr, J. A. Pesti, N. A. Magnus, J. M. Fortunak, P. N. Confalone and W. A. Nugent, Org. Lett., 2000, 2, 3119–3121 CrossRef CAS PubMed.
- Y. Yamamoto, H. Hayashi, T. Saigoku and H. Nishiyama, J. Am. Chem. Soc., 2005, 127, 10804–10805 CrossRef CAS PubMed.
- V. A. Peshkov, O. P. Pereshivko, P. A. Donets, V. P. Mehta and E. V. Van der Eycken, Eur. J. Org. Chem., 2010, 4861–4867 CrossRef CAS PubMed.
- D. S. Ermolat'ev, J. B. Bariwal, H. P. L. Steenackers, S. C. J. De Keersmaecker and E. V. Van der Eycken, Angew. Chem., Int. Ed., 2010, 49, 9465–9468 CrossRef PubMed.
- W.-J. Yoo, L. Zhao and C.-J. Li, Aldrichimica Acta, 2011, 44, 43–51 CAS.
- M. Naoi, W. Maruyama, H. Yi, Y. Akao, Y. Yamaoka and M. Shamoto-Nagai, J. Neural Transm., Suppl., 2007, 121–131 CAS.
- V. A. Peshkov, O. P. Pereshivko and E. V. Van der Eycken, Chem. Soc. Rev., 2012, 41, 3790–3807 RSC.
- M. D. Berry and A. A. Boulton, Neurotoxicol. Teratol., 2002, 24, 667–673 CrossRef CAS.
- O. Bar-Am, T. Amit, O. Weinreb, M. B. H. Youdim and S. Mandel, J. Alzheimer's Dis., 2010, 21, 361–371 CAS.
- G. Magueur, B. Crousse and D. Bonnet-Delpon, Tetrahedron Lett., 2005, 46, 2219–2221 CrossRef CAS PubMed.
- K. B. Aubrecht, M. D. Winemiller and D. B. Collum, J. Am. Chem. Soc., 2000, 122, 11084–11089 CrossRef CAS.
- T. Harada, T. Fujiwara, K. Iwazaki and A. Oku, Org. Lett., 2000, 2, 1855–1857 CrossRef CAS PubMed.
- T. Murai, Y. Mutoh, Y. Ohta and M. Murakami, J. Am. Chem. Soc., 2004, 126, 5968–5969 CrossRef CAS PubMed.
- C. Wei, Z. Li and C.-J. Li, Synlett, 2004, 1472–1483 CAS.
- L. Zani and C. Bolm, Chem. Commun., 2006, 4263–4275 RSC.
- C.-J. Li, Acc. Chem. Res., 2010, 43, 581–590 CrossRef CAS PubMed.
- V. V. Kouznetsov and L. Y. Vargas Méndez, Synthesis, 2008, 491–506 CrossRef CAS PubMed.
- A. B. Dyatkin and R. A. Rivero, Tetrahedron Lett., 1998, 39, 3647–3650 CrossRef CAS.
- R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958–2975 RSC.
- R. W. Armstrong, A. P. Combs, P. A. Tempest, S. D. Brown and T. A. Keating, Acc. Chem. Res., 1996, 29, 123–131 CrossRef CAS.
- M. J. Climent, A. Corma and S. Iborra, RSC Adv., 2012, 2, 16–58 RSC.
- B. Sreedhar, A. S. Kumar and P. S. Reddy, Tetrahedron Lett., 2010, 51, 1891–1895 CrossRef CAS PubMed.
- S. Samai, G. C. Nandi and M. S. Singh, Tetrahedron Lett., 2010, 51, 5555–5558 CrossRef CAS PubMed.
- H.-M. Ko, K. K.-Y. Kung, J.-F. Cui and M.-K. Wong, Chem. Commun., 2013, 49, 8869–8871 RSC.
- J. S. Yadav, B. V. Subba Reddy, A. V. Hara Gopal and K. S. Patil, Tetrahedron Lett., 2009, 50, 3493–3496 CrossRef CAS PubMed.
- T. He, Z. Zha, C. Pan and Z. Wang, Synth. Commun., 2007, 37, 849–858 CrossRef PubMed.
- N. P. Eagalapati, A. Rajack and Y. L. N. Murthy, J. Mol. Catal. A: Chem., 2014, 381, 126–131 CrossRef CAS PubMed.
- C. M. Wei and C.-J. Li, J. Am. Chem. Soc., 2002, 124, 5638–5639 CrossRef CAS PubMed.
- C. Koradin, K. Polborn and P. Knochel, Angew. Chem., Int. Ed. Engl., 2002, 41, 2535–2538 CrossRef CAS.
- M. Benaglia, D. Negri and G. Dell'Anna, Tetrahedron Lett., 2004, 45, 8705–8708 CrossRef CAS PubMed.
- C. Fischer and E. M. Carreira, Org. Lett., 2001, 3, 4319–4321 CrossRef CAS PubMed.
- K. Yamada and K. Tomioka, Chem. Rev., 2008, 108, 2874–2886 CrossRef CAS PubMed.
- W. Fan and S. Ma, Chem. Commun., 2013, 49, 10175–10177 RSC.
- R. Gurubrahamam and M. Periasamy, J. Org. Chem., 2013, 78, 1463–1470 CrossRef CAS PubMed.
- C.-J. Li and C. Wei, Chem. Commun., 2002, 268–269 RSC.
- J. B. Bariwal, D. S. Ermolat'ev and E. V. Van der Eycken, Chem.–Eur. J., 2010, 16, 3281–3284 CrossRef CAS PubMed.
- L. Shi, Y.-Q. Tu, M. Wang, F.-M. Zhang and C.-A. Fan, Org. Lett., 2004, 6, 1001–1003 CrossRef CAS PubMed.
- N. Guo and J.-X. Ji, Tetrahedron Lett., 2012, 53, 4797–4801 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- R. A. Sheldon, Pure Appl. Chem., 2000, 72, 1233–1246 CrossRef CAS.
- R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2013, 17, 1479–1484 CrossRef CAS.
- K. Sanderson, Nature, 2011, 469, 18–20 CrossRef CAS PubMed.
- P. J. Dunn, Chem. Soc. Rev., 2012, 41, 1452–1461 RSC.
- X. Xiong, H. Chen and R. Zhu, Catal. Commun., 2014, 54, 94–99 CrossRef CAS PubMed.
- B. Karimi, M. Gholinejad and M. Khorasani, Chem. Commun., 2012, 48, 8961–8963 RSC.
- D. Bhuyan, M. Saikia and L. Saikia, Catal. Commun., 2015, 58, 158–163 CrossRef CAS PubMed.
- B. J. Borah, S. J. Borah, L. Saikia and D. K. Dutta, Catal. Sci. Technol., 2014, 4, 1047–1054 CAS.
- J. Dulle, K. Thirunavukkarasu, M. C. Mittelmeijer-Hazeleger, D. V. Andreeva, N. R. Shiju and G. Rothenberg, Green Chem., 2013, 15, 1238–1243 RSC.
- N. Salam, A. Sinha, A. S. Roy, P. Mondal, N. R. Jana and Sk M. Islam, RSC Adv., 2014, 4, 10001–10012 RSC.
- Y. Ju, C.-J. Li and R. S. Varma, QSAR Comb. Sci., 2004, 23, 891–894 CAS.
- J. Yang, P. Li and L. Wang, Catal. Commun., 2012, 27, 58–62 CrossRef PubMed.
- P. Li, S. Regati, H.-C. Huang, H. D. Arman, B.-L. Chen and J. C.-G. Zhao, Chin. Chem. Lett., 2015, 26, 6–10 CrossRef CAS PubMed.
- M. Jeganathan, A. Dhakshinamoorthy and K. Pitchumani, ACS Sustainable Chem. Eng., 2014, 2, 781–787 CrossRef CAS.
- R. Mallampati and S. Valiyaveettil, ACS Sustainable Chem. Eng., 2014, 2, 855–859 CrossRef CAS.
- X. Xiong, H. Chen and R. Zhu, Chin. J. Catal., 2014, 35, 2006–2013 CrossRef CAS.
- G. Bosica, J. Spiteri and C. Borg, Tetrahedron, 2014, 70, 2449–2454 CrossRef CAS PubMed.
- R. Ballini, G. Bosica, A. Palmieri, F. Pizzo and L. Vaccaro, Green Chem., 2008, 10, 541–544 RSC.
- R. Maggi, G. Bosica, S. Gherardi, C. Oro and G. Sartori, Green Chem., 2005, 7, 182–184 RSC.
- M. Kidwai and A. Jahan, J. Iran. Chem. Soc., 2011, 8, 462–469 CrossRef CAS.
- T. Fukuyama, M. Shinmen, S. Nishitani, M. Sato and I. Ryu, Org. Lett., 2002, 4, 1691–1694 CrossRef CAS PubMed.
- Y. Liang, Y.-X. Xie and J.-H. Li, J. Org. Chem., 2006, 71, 379–381 CrossRef CAS PubMed.
- J. T. Guan, T. Q. Weng, G.-A. Yu and S. H. Liu, Tetrahedron Lett., 2007, 48, 7129–7133 CrossRef CAS PubMed.
- C. Girard, E. Onen, M. Aufort, S. Beauvière, E. Samson and J. Herscovici, Org. Lett., 2006, 8, 1689–1692 CrossRef CAS PubMed.
- M. Keshavarz, N. Iravani, A. Ghaedi, A. Z. Ahmady, M. Vafaei-Nezhad and S. Karimi, SpringerPlus, 2013, 2, 1–8 CrossRef PubMed.
- M. J. Albaladejo, F. Alonso, Y. Moglie and M. Yus, Eur. J. Org. Chem., 2012, 3093–3104 CrossRef CAS PubMed.
- A. Fodor, A. Kiss, N. Debreczeni, Z. Hell and I. Gresits, Org. Biomol. Chem., 2010, 8, 4575–4581 CAS.
- B. S. Navale and R. G. Bhat, RSC Adv., 2013, 3, 5220–5226 RSC.
- M. Lakshmi Kantam, S. Laha, J. Yadav and S. Bhargava, Tetrahedron Lett., 2008, 49, 3083–3086 CrossRef PubMed.
- L.-H. Zou, A. J. Johansson, E. Zuidema and C. Bolm, Chem.–Eur. J., 2013, 19, 8144–8152 CrossRef CAS PubMed.
- D. C. Owsley and C. E. Castro, Org. Synth., 1972, 52, 128–131 CrossRef CAS.
- H. Feng, D. S. Ermolat'ev, G. Song and E. V. Van der Eycken, J. Org. Chem., 2011, 76, 7608–7613 CrossRef CAS PubMed.
- M.-T. Chen and O. Navarro, Synlett, 2013, 1190–1192 CAS.
- M. B. Gawande, P. S. Branco, I. D. Nogueira, C. A. A. Ghumman, N. Bundaleski, A. Santos, O. M. N. D. Teodoro and R. Luque, Green Chem., 2013, 15, 682–689 RSC.
- E. Ramu, R. Varala, N. Sreelatha and S. R. Adapa, Tetrahedron Lett., 2007, 48, 7184–7190 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra05546f |
| This journal is © The Royal Society of Chemistry 2015 |