
Modulation of photovoltage in mesoscopic perovskite solar cell by controlled interfacial electron injection
Abstract
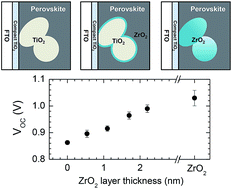
Controlled interfacial electron injection was investigated by surface modification of mesoporous TiO2 film with insulating thin ZrO2 layer in a mesoscopic CH3NH3PbI3 perovskite solar cell. Open-circuit voltage (Voc) was linearly increased with increasing the thickness of the surface ZrO2 film from 863 mV for the bare TiO2 to 988 mV for the surface modified TiO2 with 2.2 nm-thick ZrO2 layer (Voc was 1030 mV for the mesoporous ZrO2), which was due to decrease in electron injection by thickening the surface ZrO2 layer. Change in electron injection was confirmed by photoluminescence spectra. Charge collection was diminished with the surface ZrO2 layer, which might be related to the limited electron diffusion length in the solution-processed perovskite layer due to the reduced injection into TiO2. Fill factor was reproducibly improved by surface modification, which is due to increased shunt resistance. By the controlled electron injection method, a power conversion efficiency was improved from 11.7% to 13.6% by introducing 1.1 nm-thick ZrO2 layer on the TiO2 surface.
 Please wait while we load your content...
Please wait while we load your content...

