
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designing temperature-memory effects in semicrystalline polyurethane†
Nikolaus
Mirtschin
and
Thorsten
Pretsch
*
BAM Federal Institute for Materials Research and Testing, Division 6.5, Polymers in Life Science and Nanotechnology, Unter den Eichen 87, 12205 Berlin, Germany. E-mail: thorsten.pretsch@bam.de; Fax: +49 30 8104 1617; Tel: +49 30 8104 3804
First published on 22nd May 2015
Abstract
Temperature-memory polymers are able to generate a substantial mechanical response when heated above the temperature, at which a preceding deformation was carried out. Here we show how to design the temperature-memory effect (TME) by thermomechanical treatment. As a model polymer, phase segregated poly(ester urethane) (PEU) containing crystallizable segments of poly(1,4-butylene adipate) (PBA) was used. For programming, strain elongation was applied at temperatures within the PBA melting transition area, before temperature holding, unloading and cooling were carried out. Upon heating under stress-free or constant strain recovery conditions, precisely set temperature-memory onsets could be witnessed. Most importantly, strain fixities and recoverabilities the same as maximum recovery stresses turned out to be controllable by strain rate and temperature holding time after deformation, while transition temperatures remained largely unaffected. The tailoring of thermoresponsiveness was structurally enabled by different PBA crystallinities in the programmed state as verified by wide-angle X-ray scattering (WAXS). The reported studies intend to design TMEs in semicrystalline polyurethanes according to user-defined needs to make this technology broadly applicable.
1. Introduction
Temperature-memory polymers (TMPs) are able to generate a substantial mechanical response when heated above that temperature, at which a preceding deformation was carried out as an essential part of thermomechanical programming.1–6 Therefore, they received a special status among shape memory polymers (SMPs) like segmented polyurethane elastomers, whose shape recovery behavior is dominated by network architecture and the processing technique, which efficiently direct the physical and morphological properties.7–22 Ideally, fine-tuning of thermally activated shape memory responses can be accomplished by programming.23,24 While programming of one-way shape memory effects commonly consists of several steps, including polymer heating above a melting or glass transition temperature, followed by deformation and cooling under constrained conditions below the crystallization or glass transition,25–28 it is simply the deformation in a phase transition region, which adds valuable temperature-memory information to a polymer. The amount of stored mechanical energy is a function of deformation temperature Td and it is converted into internal energy as a result of the reconfiguration of polymer chains and – if applied – by cooling; in turn, the triggering of the temperature-memory effect (TME) is accompanied by the release of stored mechanical energy as determined by the relaxation behavior of the macromolecules.29–31 Once a polymer is identified as “high performance” TMP, optimization in chemical structure as frequently witnessed for SMPs32,33 is no longer necessary.Today, technological potential resides for TMPs in applications like self-sufficient release systems,3 temperature sensors,34 instruments for minimally invasive surgery35 and switchable information carriers.36 Up to now, a wide range of polymer design concepts proved to be sustainable. Temperature-memory behavior could be verified for amorphous polymer systems, including polyvinyl alcohol-based nanocomposites,1,37 perfluorosulfonic acid ionomer (Nafion),34,38,39 poly(ether urethane) and radio opaque composites thereof,40 semi-interpenetrating poly(methyl methacrylate)/poly(ethylene glycol) (PMMA/PEG) networks,5,41 covalently cross-linked PMMA-poly(ε-caprolactone) (PMMA-PCL),5 miscible poly(L-lactide)/PMMA blends,42 polyamide 12 and polyamide microfibers loaded with multiwall carbon nanotubes43 and acrylate-based polymer.6 It is remarkable that only few semicrystalline polymers qualified as versatile TMPs so far. These include poly[ethylene-ran-(vinyl acetate)] copolymers with crystallizable polyethylene segments,3 poly(ester urethanes) (PEU) containing crystallizable PCL segments,35 photo-cross-linked star PCL–PEG networks4 and PEU with crystallizable poly(1,4-butylene adipate) (PBA).36
Surprisingly, studies reporting on a tailoring of temperature-memory properties by variation of programming conditions are rare. In well-designed experiments on poly(ether urethane) and its radio opaque composites, the application of higher maximum strains, for instance, gave increased recovery stresses.40 Moreover, Nafion could be identified as fruitful example for programming-assisted control of thermoresponsiveness. In this case, shape fixing was carried out in a strain- and stress-controlled manner and control over maximum recovery speed could be achieved when applying a targeted strain.34 In another significant parametric study, the free recovery behavior of amorphous acrylate-based polymer was examined.6 Herein, decreasing the heating rate was identified to increase the recovery speed versus the recovery temperature, and it was theoretically predicted that increasing the temperature holding time after deformation will decrease the recovery speed. Apart from that, Grillard et al.43 found out that fibers made of polyamide 12 loaded with multiwall carbon nanotubes (CNT-PA12 composite fibers) lose temperature-memory when slowly deformed in the course of programming or when kept for more than 10 min at elevated temperatures. The associated loss of recovery stress was explained with the relaxation of dynamic heterogeneities, which were present in the glassy domains of the examined systems. This motivated us to investigate the influence of time-dependent programming steps on the temperature-memory behavior of a semicrystalline polymer for the first time. To address this, a PEU with distinct temperature-memory properties was selected and a recently introduced method for temperature-memory onset programming36 selectively modified by varying the strain rate and temperature holding time after deformation. Once programmed, the thermoresponsiveness was investigated both under stress-free and constant strain recovery conditions. Against this background we demonstrate that a careful parameter selection constitutes a useful way to control the temperature-memory behavior of the PEU. This way an approach was followed, which goes far beyond recent attempts to set temperature-memory transitions in semicrystalline TMPs.3,4,35 Compared with CNT-PA12 composite fibers,43 widely differing programming/temperature-memory relations could be witnessed.
2. Experimental section
2.1 Materials
The herein investigated TMP was Desmopan DP 2795A SMP, which is a poly(ester urethane) (PEU) from Bayer MaterialScience AG. Samples were received as 2 mm thick injection molded plaques. The hard segment was composed of 4,4′-methylenediphenyl diisocyanate (MDI) and a 1,4-butanediol (BD) chain extender. The soft segment was built up by poly(1,4-butylene adipate) (PBA) characterized by a molecular weight of 3500 g mol−1. The thermal properties of the PBA phase including calorimetric studies and dynamic mechanical analysis were previously reported.36 Further information regarding the molecular structure of the PEU, an attenuated total reflectance-Fourier transform-infrared (ATR-FT-IR) spectrum with signal assignment and wide-angle X-ray scattering (WAXS) data, confirming the presence of crystalline PBA units in the PEU at 23 °C, were also recently supplied.36 The mean hardness of the PEU changes from approximately 92 Shore A at 23 °C to 62 Shore A at 60 °C.44 Apart from that, Desmopan DP 2795A SMP is characterized by good shape-memory properties (ESI Fig. 1†) and proved to be well-suited for the preparation of information carriers.45–482.2 Characterization methods
Thermomechanical measurements were conducted with an electromechanical testing system (Zwick/Roell Z005), which was equipped with a thermochamber (Zwick/Roell) and a temperature controller (Eurotherm 2261e). Test procedures were designed with the software testXpert® II (V 3.31). For specimen preparation, type 5B tensile bars49 were punched out of the PEU plaques. Prior usage, specimens were annealed for 10 min at 60 °C and stored for at least one week at 23 °C (50% air humidity). At the beginning of a measurement, a tensile bar was clamped with an initial gauge length of 10 mm into the pneumatic grips of the electromechanical testing system. Typically, a clamping pressure of 5.2 bar was selected. In the course of thermomechanical measurements, changes in normal force were followed with a 100 N load cell. In parallel, the stress σ was determined by dividing the force through the initial cross section of the specimen. During each measurement, changes in strain ε were followed from crosshead displacement.To program temperature-memory onsets into PEU, specimens were initially cooled or heated from 23 °C to the deformation temperature Td = −10, 0, 10, 20, 30 and 40 °C. Adjacently, different programming approaches were followed and the temperature-memory behavior was examined. In the first scenario, a maximum strain εm of 100% was applied with a strain rate ε′ of 1% min−1 or 3 × 104% min−1. In the obtained stress–strain diagram, the Young's modulus E was calculated from the initial slope as ratio of stress to strain. The yield point including the associated strain εy and stress σy was determined from the early local maximum in the stress–strain curves. In those cases where no local maximum occurred, the two tangent intersection method was used to estimate the onset of plastic deformation.50 After 5 min at Td, the specimen was unloaded with a rate of 10% min−1 before it was finally cooled to −20 °C.
In the other programming series, the temperature holding time th after deformation was varied. For this purpose, a strain rate of 3 × 102% min−1 was selected while εm was set to 100% and th to 5 min or 15 h. To finalize programming, the specimen was unloaded at Td and cooled to −20 °C, where the fixed strain εu was determined.
Independent of the programming method, thermoresponsiveness was adjacently studied under stress-free and constant strain recovery conditions. Therefore, specimens were heated from −20 to 80 °C. In case of free strain recovering, the strain εp characterizing the recovered shape was determined. Additionally, the recovered strain εu − εp was quantified. The strain fixity ratio Rf and the total strain recovery ratio Rr,tot were calculated according eqn (1) and (2):
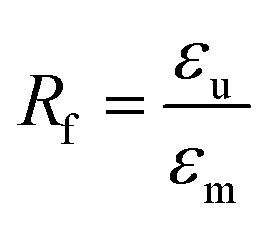 | (1) |
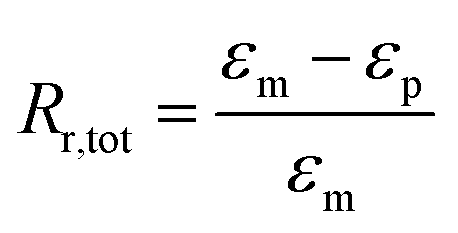 | (2) |
To precisely characterize the switching transitions, tangent lines were established in the recovery curve of the strain-temperature test protocol. The temperatures corresponding to their intersection points at the beginning and toward the end of heating were defined as on- and offset switching temperature Tsw,on and Tsw,off, respectively.
When examining the stress recovery behavior, the maximum recovery stress σmax was determined. For a detailed analysis, tangent lines were established in the stress-temperature test protocol. Those temperatures, which corresponded to their intersection points at the beginning and toward the end of heating, were defined as on- and offset stress recovery temperature Tσ,on and Tσ,off.
Infrared (IR) thermography was used to follow the temperature distribution on the surface of a PEU specimen during elongation at Td = 20 °C. For this experiment a strain rate of 3 × 104% min−1 and an elongation of 100% were selected. The temperature was recorded with a microbolometer IR camera VarioCAM® hr from InfraTec, which operated in a spectral range of 7.5–14.0 μm and was equipped with a close-up lens, which had a resolution of 50 μm. The camera was placed at a distance of less than 10 mm from the specimen. The emissivity of PEU surface was set to 1. Thermal data was recorded using a frame rate of 50 Hz and an array size of 640 × 480 pixel.
In situ wide-angle X-ray scattering (WAXS) was performed at the synchrotron microfocus beamline “μSpot” at Bessy II (Helmholtz Centre Berlin for Materials and Energy). The employed diffraction setup was identical with the one described by Paris et al.51 The experiments were carried out at a wavelength of 1 Å and an exposure time of 15 s. A two-dimensional X-ray detector (MarMosaic, CCD 3072 × 3072) was used to collect the scattered radiation at a distance of 170 mm behind the specimen. A temperature controlled sample holder unit TCS 120 (Anton Paar, Austria), equipped with a Peltier heater and a Pt 100 temperature sensor, was used for heating. Initially, a pristine PEU specimen was cut at 8 °C to a length of 13 mm and loosely attached to the precooled sample holder. Subsequently, the specimen was heated from 8 to 60 °C using a rate of 3 °C min−1. In parallel, scattering images were recorded. The computer program FIT2D52 was employed to obtain diagrams of scattered intensities versus scattering angle 2θ (with respect to the wavelength of the Cu Kα-line). The degree of PEU crystallinity χc was estimated from the obtained WAXS patterns by dividing the integrated intensity due to scattering from crystallites Icryst through the totally scattered intensity Itotal:
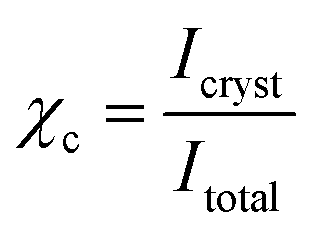 | (3) |
Furthermore, various specimens were examined by means of WAXS directly after programming (Td = 10 °C: ε′ = 1% min−1 or 3 × 104% min−1; Td = 10 or 40 °C: ε′ = 3 × 102% min−1 and th = 5 min or 15 h). Again, the same setup as described above was used and a testing temperature of 10 °C selected.
3. Results and discussion
One criterion which qualifies phase segregated, semicrystalline polymer networks like PEUs as versatile TMPs is that the soft (mobile) phase is characterized by a broad thermal transition.36 This motivated us to start our examinations with in situ WAXS on a model PEU, herein used as TMP (Fig. 1). The transition associated with the melting of the poly(1,4-butylene adipate) (PBA) phase was found to spread from 10 °C (χc = 12.2%) to 56 °C. The overall crystallinity of the PEU was dominated by the soft segment. In the WAXS patterns, only one weak signal at 2θ = 21.3° could be identified which remained unaffected by temperature variation and therefore seemed to derive from the hard segment (ESI Fig. 2†).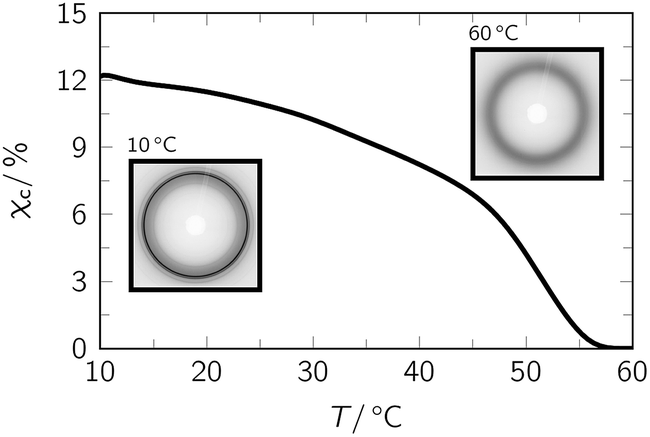 | ||
| Fig. 1 Evolution of overall crystallinity χc when heating pristine PEU during an in situ WAXS measurement. The insets show the diffractograms at the beginning and end of the measurement. | ||
Subsequently, thermomechanical treatment was applied as essential part of programming TMEs (Fig. 2). The whole programming history is exhibited in Fig. 2a. Initially, PEU specimens were tensile loaded at different deformation temperatures Td using strain rates ε′ of 1% min−1 and 3 × 104% min−1, respectively. The stress–strain behavior was followed, first of all, until specimens were unloaded (Fig. 2b–d and ESI Fig. 3†). According to the evolution of Young's modulus E, material stiffness increased at lower Td (see caption of Fig. 2). This had been expected from the rising crystallinity content in the pre-deformation states (Fig. 1). In turn, the elasticity of the material decreased at lower temperatures (ESI Fig. 3†) as suggested by a contemporaneous decrease in strain at yield (ESI Fig. 3a†) and increase in stress at yield (ESI Fig. 3b†). One can also notice that elasticity depended on strain rate as exemplified by consistently lower strains and stresses at yield when applying slow deformation (ESI Fig. 3†). Further, it is apparent that the tensile stress required to elongate the specimens to εm = 100% systematically increased at lower Td. In this context, a steady growth in deformation resistance was found in the stress–strain plots of slow deformation, which became steeper at higher elongations. This effect intensified at lower temperatures and can be explained with the occurrence of material hardening through strain-induced PBA crystallization.53 Moreover, necking took place after rapid deformation at Td ≤ 20 °C as already witnessed for other urethane-based polymers.54,55 Since neck formation and propagation in polymers can be accompanied with heat generation56–58 – the interrelation of temperature rise with stretching was discovered by Gough when studying caoutchouc59 – we decided to extend our investigations toward infrared thermography by exemplarily following the specimen temperature at its surface during elongation at 20 °C (Fig. 3). With regard to the obtained image series it can be seen that two necks developed simultaneously at a strain of about 40% and thus beyond the maximum of the corresponding stress–strain curve (see dotted, pastel green line in Fig. 2b). The observed behavior is commonly favored in polymers by high extension rates and sometimes referred to secondary or double necking.60 In the course of rapid elongation, portion of the work expended on deformation started transforming into heat. Shortly after finalizing the deformation process, the highest temperature was recorded in the necking regions, illustrating a temperature increase with reference to the specimen's pre-deformation state by approximately 6 °C.
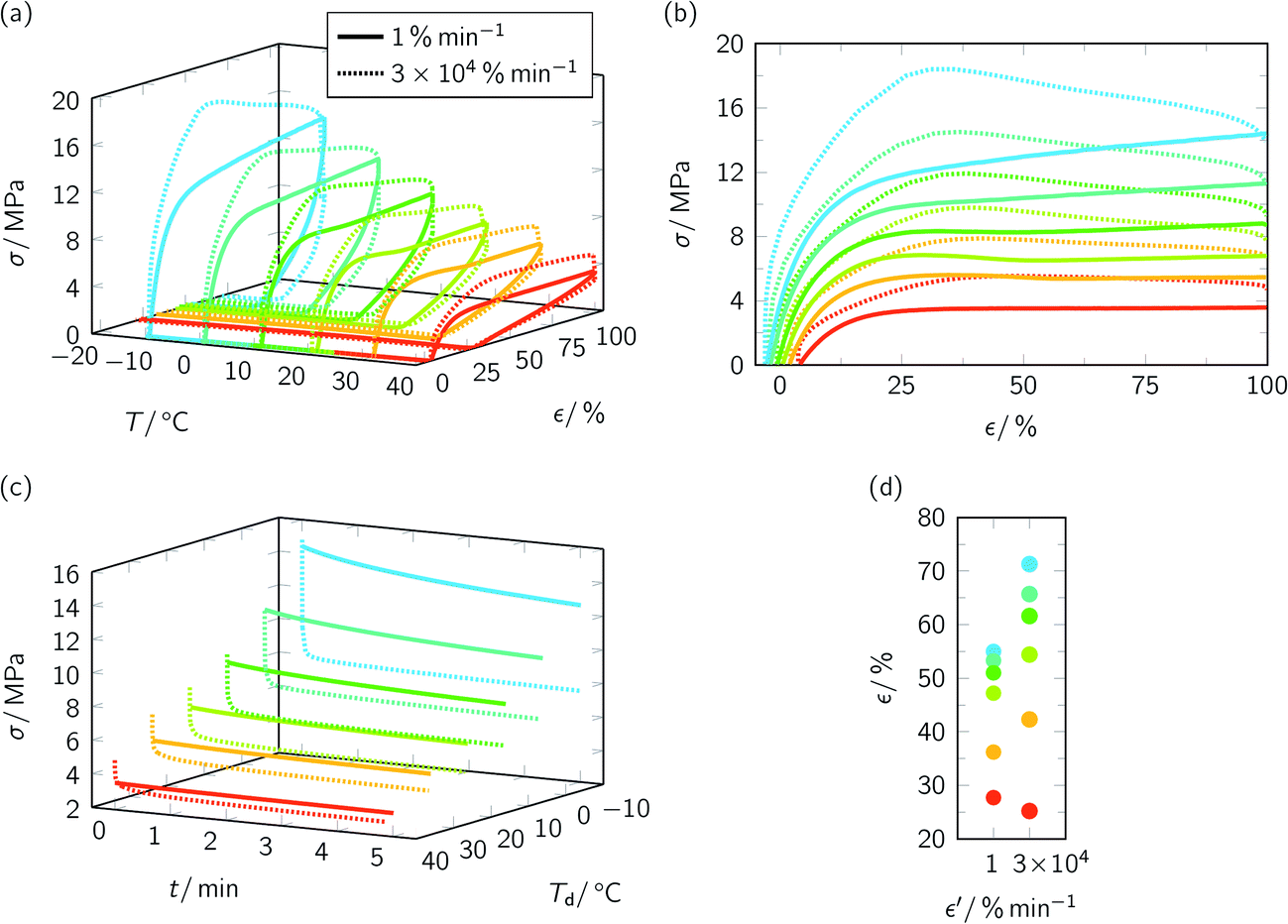 | ||
| Fig. 2 Programming of various temperature-memory effects in PEU. Stress–strain-temperature protocol (a) and diagrams showing the stress–strain behavior during deformation ((b), strain rate ε′ = 1% min−1 and 3 × 104% min−1), stress relaxation behavior when keeping the specimens at εm = 100% (c) and nominal strain after unloading with 10% min−1 at Td (d). For ε′ = 1% min−1, Young's moduli E of 166 MPa (Td = −10 °C), 131 MPa (Td = 0 °C), 114 MPa (Td = 10 °C), 96 MPa (Td = 20 °C), 78 MPa (Td = 30 °C) and 43 MPa (Td = 40 °C) were determined. | ||
 | ||
| Fig. 3 Sequence of thermographic images recorded at 20 °C during elongation (ε′ = 3 × 104% min−1, (a–f)) and 1.1 s after stretching (g) a PEU specimen. The corresponding strain values were 0% (a), 20% (b), 40% (c), 60% (d), 80% (e) and 100% (f and g). The formation of an upper and a lower neck is illustrated by the arrows. | ||
Anyway, to continue with the programming of TMEs, loaded specimens were exposed to a 5 min temperature holding step at Td while the elongation was maintained. During this time, stress relaxed significantly, but less strongly when loading was carried out with the lower strain rate (Fig. 2c). The reason is that PBA crystals, which derived from the pre-deformation state, and further PBA crystals, which formed in the course of elongation (a sufficient thermal stability is assumed), counteracted the recovering of the specimens. By contrast, much stronger stress declines were witnessed during temperature holding subsequent to rapid deformation (Fig. 2c), culminating in lower stresses before unloading (ESI Fig. 4†). Obviously, some PBA crystals, which were present in the pre-deformation states, were molten and could no longer serve as physical cross-links. However, we conjecture that briefly after deformation significant crystallization occurred at Td. This could explain the in tendency higher strains after unloading compared with those detected after slow deformation (Fig. 2d). To prove our assumption, WAXS measurements were carried out on programmed PEU, which was deformed at 10 °C with slow and high strain rate while the other thermomechanical parameters remained unchanged (ESI Fig. 5†). The analysis of the WAXS diffractograms gave evidence that strain rate had an impact on the overall crystallinity χc. When deforming the specimen more rapidly (ε′ = 3 × 104% min−1), a χc of 20.5% was determined; in turn, it was 17.2% when choosing a lower strain rate (ε′ = 1% min−1). To further confirm that time-dependent programming steps were affecting the thermomechanical behavior, the total time at which specimens were kept at Td was varied by modifying the unloading rate, and the influence on residual strains was determined. As a matter of fact, a Td of −10 °C and a strain rate of 3 × 104% min−1 were selected, the temperature holding step after deformation was omitted and specimens were unloaded with different rates before they were cooled to −20 °C. In accordance with the above obtained picture, it became obvious that nominal strain gradually increased with decelerating unloading rate and thus with progressing storage time at Td (1 × 104% min−1: 43%; 3 × 103% min−1: 45%; 3 × 102% min−1: 56%; 10% min−1: 66%). Again, growing PBA crystallinity seemingly supported a remarkable enhancement in fixed strain.
To study the influence of strain rate on temperature-memory behavior under stress-free and constant strain recovery conditions, the specimens whose thermomechanical treatment is shown in Fig. 2 were heated to 80 °C (Fig. 4, Table 1). Taking a closer look at the data illustrates that thermomechanical properties were largely depending on programming. At the 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times higher strain rate, progressive PBA crystallization after specimen drawing substantiated a higher strain fixity ratio Rf (Table 1). Under stress-free recovery conditions, larger fixed strain inevitably resulted in enhanced recovery strain εu − εp, which impressively demonstrates an adjustability of the most relevant thermomechanical parameters through strain rate variation. This way, it could be shown that manipulation of the stress-free recovery behavior of TMPs is not restricted to the variation of programming parameters like εm40 or cooling conditions after deformation.6,34 However, an exception was the Td = 40 °C scenario (Table 1, Fig. 4a, above). Here, the PBA phase had its lowest pre-deformation crystallinity and no clear influence of strain rate on temperature-memory properties could be found. Regarding the maximum recovery stress σmax, the influence of strain rate was almost negligible for Td = 30 and 40 °C. In contrast, systematically lower σmax values were determined upon rapid deformation for Td ≤ 20 °C (Table 1, Fig. 4a, below). In the first instance, this behavior was unexpected, because PBA crystallinity seemed to be higher, and motivated us to study the recovery of the necks. Intriguingly, local reversion of the necking processes could be observed both under stress-free and constant strain recovery conditions for specimens, which were deformed at −10 °C (Fig. 5). As visible to the naked eye, neck recovering was finalized under stress-free conditions somewhere in the middle of the switching transition (Fig. 5a, also compare Fig. 4a). In turn, the upper and lower neck recovered under constant strain conditions at a significantly higher temperature, which was well above Tσ,max and close to Tσ,off (Table 1, Fig. 5b). Since no necking could be detected after slow elongation, the necks observed after rapid elongation at Td ≤ 20 °C and their slow recovering give a reasonable explanation for a lowering in maximum recovery stress (Table 1, Fig. 4a, below). Nevertheless, satisfying the criterion of temperature-memory behavior, transition temperatures (Tsw,on, Tσ,on) increased linearly with Td (Fig. 4b) and the temperature regime of responsiveness remained unaffected by strain rate. Furthermore, temperatures corresponding to σmax did not shift when varying the strain rate (Fig. 4a, below). This way, a more stable material behavior could be detected for PEU compared with CNT-PA12 temperature-memory fibers, for which a remarkable loss and shift in recovery stress were verified after slow deformation.43 However, the temperatures, at which strain and stress finally stabilized (Fig. 4a), corresponded to the recently determined DSC offset melting temperature of the PBA phase36 and were in analogy with the in situ WAXS data of untreated PEU (Fig. 1). Throughout the measurement series, the recovered strain εu − εp and the maximum recovery stress σmax declined at higher Td due to the increasing elasticity of the TMP at higher temperatures. In turn, the total strain recovery ratio Rr,tot remained permanently around 90%.
000 times higher strain rate, progressive PBA crystallization after specimen drawing substantiated a higher strain fixity ratio Rf (Table 1). Under stress-free recovery conditions, larger fixed strain inevitably resulted in enhanced recovery strain εu − εp, which impressively demonstrates an adjustability of the most relevant thermomechanical parameters through strain rate variation. This way, it could be shown that manipulation of the stress-free recovery behavior of TMPs is not restricted to the variation of programming parameters like εm40 or cooling conditions after deformation.6,34 However, an exception was the Td = 40 °C scenario (Table 1, Fig. 4a, above). Here, the PBA phase had its lowest pre-deformation crystallinity and no clear influence of strain rate on temperature-memory properties could be found. Regarding the maximum recovery stress σmax, the influence of strain rate was almost negligible for Td = 30 and 40 °C. In contrast, systematically lower σmax values were determined upon rapid deformation for Td ≤ 20 °C (Table 1, Fig. 4a, below). In the first instance, this behavior was unexpected, because PBA crystallinity seemed to be higher, and motivated us to study the recovery of the necks. Intriguingly, local reversion of the necking processes could be observed both under stress-free and constant strain recovery conditions for specimens, which were deformed at −10 °C (Fig. 5). As visible to the naked eye, neck recovering was finalized under stress-free conditions somewhere in the middle of the switching transition (Fig. 5a, also compare Fig. 4a). In turn, the upper and lower neck recovered under constant strain conditions at a significantly higher temperature, which was well above Tσ,max and close to Tσ,off (Table 1, Fig. 5b). Since no necking could be detected after slow elongation, the necks observed after rapid elongation at Td ≤ 20 °C and their slow recovering give a reasonable explanation for a lowering in maximum recovery stress (Table 1, Fig. 4a, below). Nevertheless, satisfying the criterion of temperature-memory behavior, transition temperatures (Tsw,on, Tσ,on) increased linearly with Td (Fig. 4b) and the temperature regime of responsiveness remained unaffected by strain rate. Furthermore, temperatures corresponding to σmax did not shift when varying the strain rate (Fig. 4a, below). This way, a more stable material behavior could be detected for PEU compared with CNT-PA12 temperature-memory fibers, for which a remarkable loss and shift in recovery stress were verified after slow deformation.43 However, the temperatures, at which strain and stress finally stabilized (Fig. 4a), corresponded to the recently determined DSC offset melting temperature of the PBA phase36 and were in analogy with the in situ WAXS data of untreated PEU (Fig. 1). Throughout the measurement series, the recovered strain εu − εp and the maximum recovery stress σmax declined at higher Td due to the increasing elasticity of the TMP at higher temperatures. In turn, the total strain recovery ratio Rr,tot remained permanently around 90%.
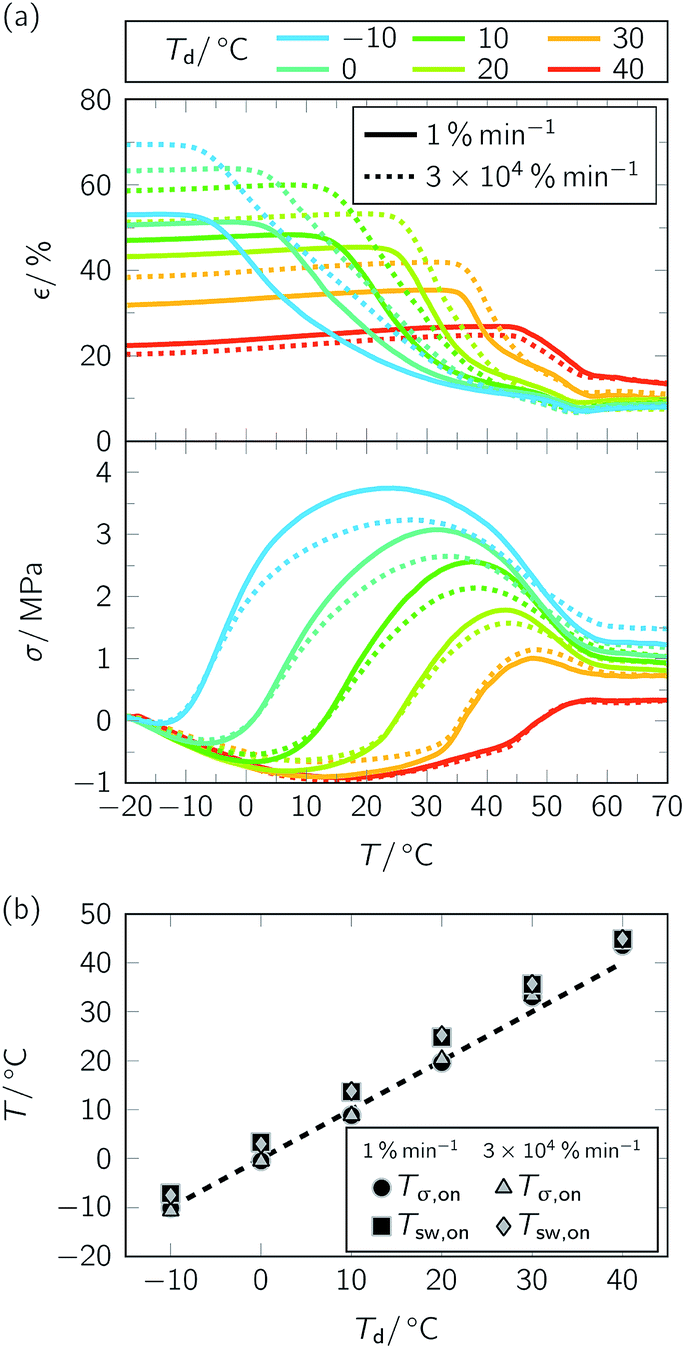 | ||
| Fig. 4 Influence of strain rate ε′ on temperature-memory behavior of PEU (εm = 100%). Consideration of ε′ = 1% min−1 (solid lines) and ε′ = 3 × 104% min−1 (dotted lines). Results of stress-free and constant strain recovery measurements (a) and evolution of Tsw,on and Tσ,on with Td (b). | ||
| T d (°C) | ε′ (% min−1) | R f (%) | Stress-free recovery conditions | Constant strain recovery conditions | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R r,tot (%) | ε u − εp (%) | T sw,on (°C) | T sw,off (°C) | T σ,on (°C) | T σ,max (°C) | T σ,off (°C) | σ max (MPa) | |||
| −10 | 1 | 53 ± 2 | 91 ± 2 | 44 ± 2 | −7 ± 1 | 55 ± 2 | −10 ± 2 | 24 ± 2 | 57 ± 2 | 3.7 ± 0.2 |
| 0 | 1 | 51 ± 2 | 91 ± 2 | 42 ± 2 | 3 ± 1 | 55 ± 2 | −1 ± 2 | 32 ± 2 | 56 ± 2 | 3.1 ± 0.2 |
| 10 | 1 | 47 ± 2 | 91 ± 2 | 38 ± 2 | 14 ± 1 | 55 ± 2 | 9 ± 2 | 37 ± 2 | 57 ± 2 | 2.6 ± 0.2 |
| 20 | 1 | 43 ± 2 | 90 ± 2 | 33 ± 2 | 25 ± 1 | 54 ± 2 | 20 ± 2 | 43 ± 2 | 57 ± 2 | 1.8 ± 0.2 |
| 30 | 1 | 32 ± 2 | 91 ± 2 | 23 ± 2 | 36 ± 1 | 55 ± 2 | 33 ± 2 | 48 ± 2 | 58 ± 2 | 1.0 ± 0.2 |
| 40 | 1 | 22 ± 2 | 88 ± 2 | 10 ± 2 | 45 ± 1 | 57 ± 2 | 44 ± 2 | 57 ± 2 | 61 ± 2 | 0.3 ± 0.2 |
| −10 | 3 × 104 | 70 ± 2 | 92 ± 2 | 62 ± 2 | −8 ± 1 | 53 ± 2 | −11 ± 2 | 27 ± 2 | 59 ± 2 | 3.2 ± 0.2 |
| 0 | 3 × 104 | 63 ± 2 | 92 ± 2 | 55 ± 2 | 3 ± 1 | 52 ± 2 | −1 ± 2 | 34 ± 2 | 58 ± 2 | 2.7 ± 0.2 |
| 10 | 3 × 104 | 59 ± 2 | 90 ± 2 | 49 ± 2 | 14 ± 1 | 53 ± 2 | 9 ± 2 | 38 ± 2 | 58 ± 2 | 2.1 ± 0.2 |
| 20 | 3 × 104 | 51 ± 2 | 92 ± 2 | 43 ± 2 | 25 ± 1 | 54 ± 2 | 20 ± 2 | 43 ± 2 | 59 ± 2 | 1.6 ± 0.2 |
| 30 | 3 × 104 | 38 ± 2 | 89 ± 2 | 27 ± 2 | 36 ± 1 | 55 ± 2 | 33 ± 2 | 48 ± 2 | 59 ± 2 | 1.2 ± 0.2 |
| 40 | 3 × 104 | 20 ± 2 | 87 ± 2 | 7 ± 2 | 45 ± 1 | 56 ± 2 | 44 ± 2 | 57 ± 2 | 61 ± 2 | 0.3 ± 0.2 |
 | ||
| Fig. 5 Thermoresponsiveness of programmed PEU specimens (Td = −10 °C, ε′ = 3 × 104% min−1, εm = 100%) under stress-free recovery conditions (a) and constant strain recovery conditions (b). The position of the upper and lower neck is illustrated by the arrows. | ||
In another attempt to gain control over the TME, the temperature holding time th at Td was drastically extended. Similar as above, we started our examinations with the stress–strain behavior, but used a strain rate ε′ of 3 × 102% min−1 (Fig. 6 and ESI Fig. 6 and 7†). Once again, the whole programming history is exhibited in a stress–strain-temperature diagram (Fig. 6a). In essence, the tensile deformation behavior (Fig. 6b) was similar as expected. First of all, values for strain and stress at yield (ESI Fig. 6†) were in between those which could be determined when selecting a slower and a higher stain rate, respectively (ESI Fig. 3†). Secondly, as can be seen in Fig. 6c, an extension of temperature holding time at Td from 5 min to 15 h caused proceeding stress relaxation, which was more pronounced at lower temperatures (ESI Fig. 7†) and seemed to be accompanied by PBA crystallization. From programming one-way shape-memory effects in segmented polyurethanes it is well-known that elongated specimens show pronounced stress decline, which goes along with significant crystallization of the soft segment phase.61–64 To analyze the influence of temperature holding time at Td on PBA crystallinity, further WAXS measurements were carried out (ESI Fig. 8†). The experiments unveiled that drastic extension of th favored an increase in overall crystallinity χc from 18.6% to 18.9% for Td = 10 °C (ESI Fig. 8a†) and from 17.1% to 22.2% for Td = 40 °C (ESI Fig. 8b†). These higher fractions of stiff crystalline regions probably caused a reduction of chain mobility and therefore much higher strains after unloading (Fig. 6d). However, in agreement with the other measurement series (Fig. 4a), specimens exhibited distinct temperature-memory behavior when heated to 80 °C as proven under stress-free and constant strain recovery conditions (Fig. 7, Table 2). As apparent in all measurements, the extension in th supported improved strain fixities (Rf ratios) and strain recoverabilities (εu − εp) while the impact on Rr,tot remained negligible (Table 2, Fig. 7a, above). Nominally, significant crystallization of the switching segment at 40 °C (ESI Fig. 8†), for instance, led to gains in Rf ratio by 39% and in strain recoverability by 36%. In this sense, isothermal crystallization turned useful to enhance control both over the fixed and recovered strain, even at a temperature, at which strain rate variation remained without impact. Since the experimental findings were always based on the same maximum strain applied (εm = 100%) and due the fact that the programming route allowed for temperature-memory onset control (Fig. 7a),36 an unprecedented fine-tuning of thermoresponsiveness could be achieved for a TMP under stress-free recovery conditions. Under constant strain recovery conditions, preceding PBA crystallization during programming counteracted specimen relaxation, thus favoring substantially superior maximum recovery stresses σmax in the ensuing heating run (Fig. 7a, below). A similar trend, namely a rise in contracting force with growing crystallinity content, is known from structurally related polyurethane-based shape memory polymers.11,22,65 Thus, extension of th at Td supported a different material behavior compared to CNT-PA12 composite fibers, for which ongoing storage time at Td resulted in a decrease in σmax, culminating in an entire loss of temperature-memory behavior.43 Incidentally, a σmax of 4.7 MPa was recorded for the PEU when deformed at −10 °C; it was the highest maximum recovery stress compared with the other semicrystalline TMPs investigated so far.3,35
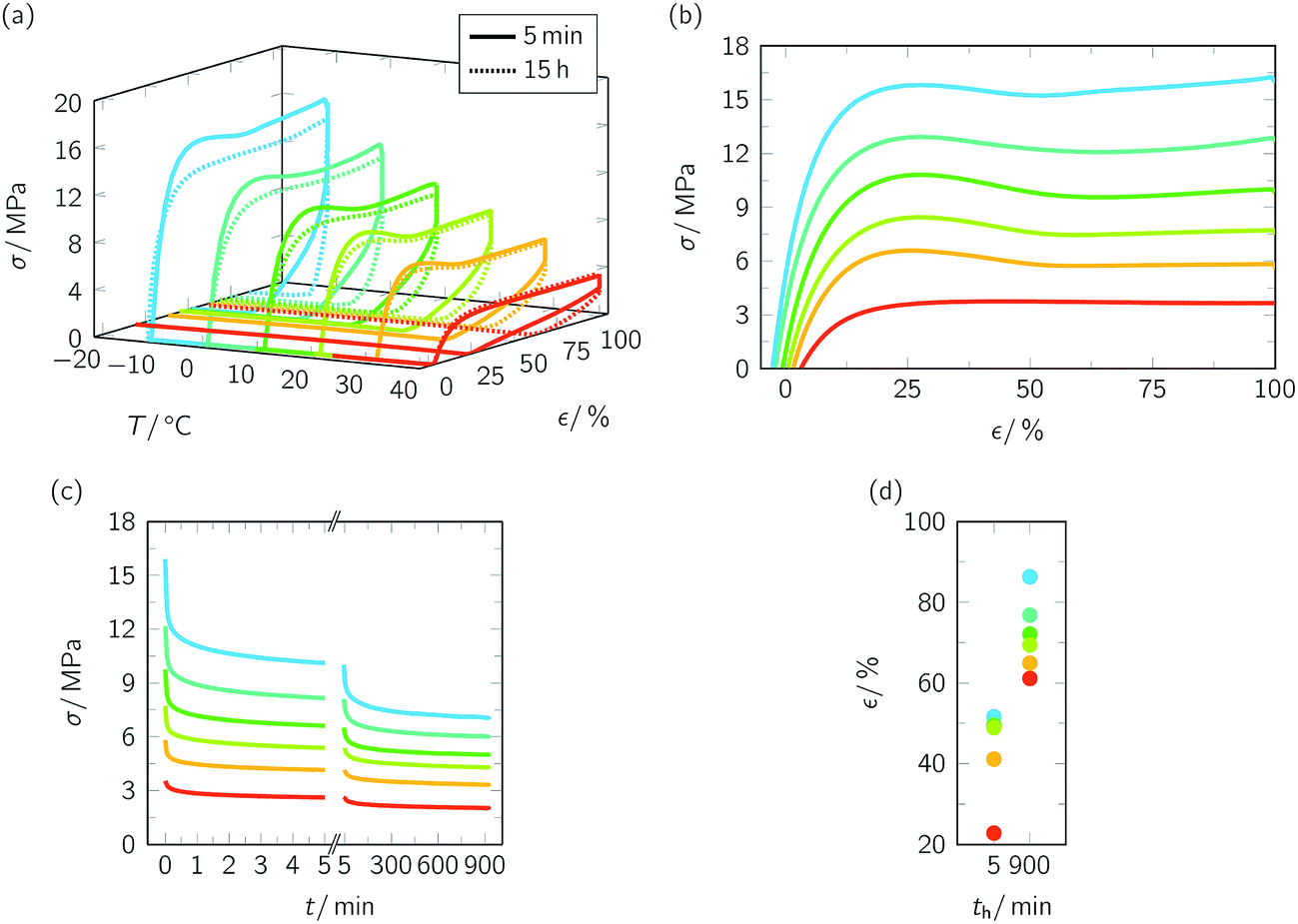 | ||
| Fig. 6 Programming of various temperature-memory effects in PEU. Stress–strain-temperature protocol (a) and diagrams showing the stress–strain behavior during deformation ((b), ε′ = 3 × 102% min−1), stress relaxation behavior when keeping the specimens at εm = 100% ((c), th = 5 min and 15 h) and nominal strain after unloading with 10% min−1 at Td (d). The obtained Young's moduli E were 217 MPa (Td = −10 °C), 156 MPa (Td = 0 °C), 144 MPa (Td = 10 °C), 107 MPa (Td = 20 °C), 97 MPa (Td = 30 °C) and 48 MPa (Td = 40 °C). | ||
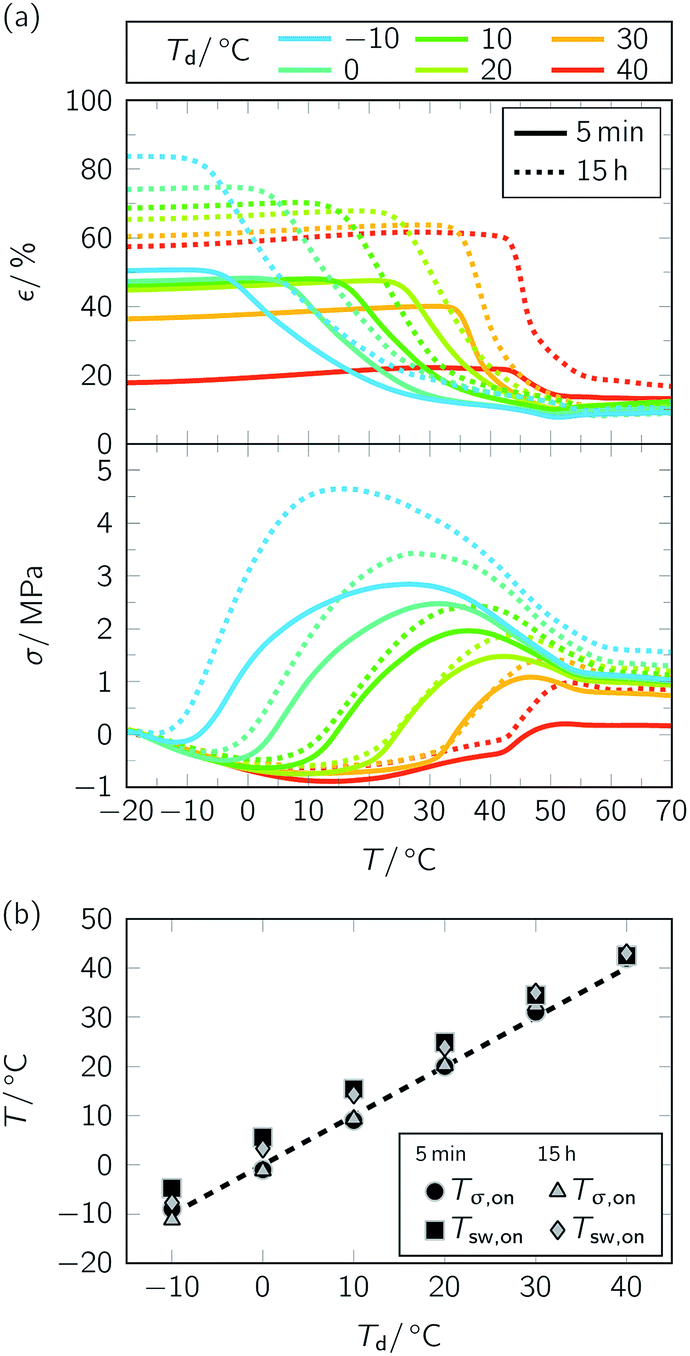 | ||
| Fig. 7 Influence of temperature holding time th at Td (ε = 3 × 102% min−1) on temperature-memory behavior of PEU. Consideration of 5 min ((a), solid lines; data were taken from a recent publication36) and 15 h ((a), dotted lines). Results of stress-free and constant strain recovery measurements (a) and evolution of Tsw,on and Tσ,on with Td (b). | ||
| T d (°C) | t h (min) | R f (%) | Stress-free recovery conditions | Constant strain recovery conditions | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R r,tot (%) | ε u − εp (%) | T sw,on (°C) | T sw,off (°C) | T σ,on (°C) | T σ,max (°C) | T σ,off (°C) | σ max (MPa) | |||
| −10 | 5 | 50 ± 2 | 90 ± 3 | 40 ± 3 | −5 ± 1 | 49 ± 3 | −9 ± 2 | 27 ± 2 | 54 ± 2 | 2.8 ± 0.2 |
| 0 | 5 | 47 ± 2 | 89 ± 3 | 36 ± 3 | 6 ± 1 | 48 ± 3 | −1 ± 2 | 32 ± 2 | 54 ± 2 | 2.5 ± 0.2 |
| 10 | 5 | 46 ± 2 | 87 ± 3 | 33 ± 3 | 15 ± 1 | 49 ± 3 | 9 ± 2 | 36 ± 2 | 54 ± 2 | 2.0 ± 0.2 |
| 20 | 5 | 45 ± 2 | 88 ± 3 | 33 ± 3 | 25 ± 1 | 50 ± 3 | 20 ± 2 | 43 ± 2 | 54 ± 2 | 1.5 ± 0.2 |
| 30 | 5 | 36 ± 2 | 88 ± 3 | 24 ± 3 | 35 ± 1 | 49 ± 3 | 31 ± 2 | 47 ± 2 | 55 ± 2 | 1.1 ± 0.2 |
| 40 | 5 | 18 ± 2 | 87 ± 3 | 5 ± 3 | 43 ± 1 | 50 ± 3 | 42 ± 2 | 52 ± 2 | 56 ± 2 | 0.2 ± 0.2 |
| −10 | 900 | 84 ± 3 | 89 ± 2 | 73 ± 3 | −8 ± 1 | 54 ± 2 | −11 ± 1 | 15 ± 2 | 58 ± 2 | 4.7 ± 0.2 |
| 0 | 900 | 74 ± 3 | 91 ± 2 | 65 ± 3 | 3 ± 1 | 57 ± 2 | −1 ± 1 | 28 ± 2 | 58 ± 2 | 3.4 ± 0.2 |
| 10 | 900 | 69 ± 3 | 89 ± 2 | 58 ± 3 | 14 ± 1 | 56 ± 2 | 9 ± 1 | 39 ± 2 | 59 ± 2 | 2.4 ± 0.2 |
| 20 | 900 | 65 ± 3 | 89 ± 2 | 54 ± 3 | 24 ± 1 | 55 ± 2 | 20 ± 1 | 44 ± 2 | 59 ± 2 | 1.9 ± 0.2 |
| 30 | 900 | 60 ± 3 | 90 ± 2 | 50 ± 3 | 35 ± 1 | 56 ± 2 | 32 ± 1 | 50 ± 2 | 59 ± 2 | 1.4 ± 0.2 |
| 40 | 900 | 57 ± 3 | 84 ± 2 | 41 ± 3 | 43 ± 1 | 56 ± 2 | 43 ± 1 | 53 ± 2 | 60 ± 2 | 1.0 ± 0.2 |
Finally, it is worth underlining that variation of temperature holding time did not significantly impart the transition temperatures. Same as in the first measurement series (Fig. 4b), recovery temperatures Tsw,on and Tσ,on increased almost linearly with Td over the whole temperature range investigated (Fig. 7b). In contrast to TMPs, whose switching segments are vitrified during programming and for which relaxation effects may lead to a loss of temperature-memory information as exemplified by an increase in strain recovery temperature with ongoing temperature holding time at Td,6 strain fixing through crystallization did not shift the recovery temperature. Thus, the shape fixing and switching mechanism proved once more to be reliable.
4. Conclusions
Modification of programming parameters like strain rate and temperature holding time at Td provided a facile strategy to gain precise control over the temperature-memory behavior of semicrystalline polyurethane. Slow loading favored strain-induced crystallization of the switching segment, rapid deformation resulted in necking, specimen heating and significant crystallization. Both allowed designing TMEs under stress-free recovery conditions. Although identified as thermoreversible phenomenon, necking seemed to account for a slight reduction in maximum recovery stress. Apart from that, it became clear that not all TMPs lose memory with age; this behavior seems rather to be limited to the amorphous ones.43 In particular, the extension of temperature holding time at Td did not allow temperature-memory amnesia for semicrystalline TMP, but was the key to strongly improve the material behavior. The associated gain in crystallinity enabled significantly higher strain fixities, strain recoverabilities and maximum recovery stresses. Basically, programming-assisted tailoring of TMEs can also be understood as tool to optimize material behavior according user-defined needs. This could stimulate innovation, e.g. in applications like thermo-sensitive grippers, sensing elements and information carriers.Acknowledgements
The research has been carried out with the support of the German Federal Ministry of Education and Research (BMBF, project funding reference number 16V0043). N. M. thanks Manfred Wagner for fruitful discussions. The authors express their gratitude to Maren Erdmann for carrying out thermomechanical measurements, Franziska Emmerling and Ralf Bienert for their advice and help regarding the WAXS measurements and Christiane Maierhofer and Mathias Röllig for IR thermographic measurements. The authors acknowledge Bayer MaterialScience AG for kindly providing the PEU.References
- P. Miaudet, A. Derré, M. Maugey, C. Zakri, P. M. Piccione, R. Inoubli and P. Poulin, Science, 2007, 318, 1294–1296 CrossRef CAS PubMed.
- L. Sun and W. M. Huang, Soft Matter, 2010, 6, 4403–4406 RSC.
- K. Kratz, S. A. Madbouly, W. Wagermaier and A. Lendlein, Adv. Mater., 2011, 23, 4058–4062 CrossRef CAS PubMed.
- L. Wang, S. Di, W. Wang, H. Chen, X. Yang, T. Gong and S. Zhou, Macromolecules, 2014, 47(5), 1828–1836 CrossRef CAS.
- Y. Wang, J. Li, X. Li, Y. Pan, Z. Zheng, X. Ding and Y. Peng, RSC Adv., 2014, 4(39), 20364–20370 RSC.
- K. Yu and H. J. Qi, Soft Matter, 2014, 10(47), 9423–9432 RSC.
- B. K. Kim, S. Y. Lee and M. Xu, Polymer, 1996, 37(26), 5781–5793 CrossRef CAS.
- F. K. Li, J. N. Hou, W. Zhu, X. Zhang, M. Xu, X. L. Luo, D. Z. Ma and B. K. Kim, J. Appl. Polym. Sci., 1996, 62(4), 631–638 CrossRef CAS.
- F. Li, X. Zhang, J. Hou, M. Xu, X. Luo, D. Ma and B. K. Kim, J. Appl. Polym. Sci., 1997, 64(8), 1511–1516 CrossRef CAS.
- B. S. Lee, B. C. Chun, Y.-C. Chung, K. I. Sul and J. W. Cho, Macromolecules, 2001, 34, 6431–6437 CrossRef CAS.
- P. Ping, W. Wang, X. Chen and X. Jing, Biomacromolecules, 2005, 6, 587–592 CrossRef PubMed.
- W. Wang, P. Ping, X. Chen and X. Jing, Eur. Polym. J., 2006, 42(6), 1240–1249 CrossRef CAS.
- S. Chen, J. Hu, Y. Liu, H. Liem, Y. Zhu and Y. Liu, J. Polym. Sci., Part B: Polym. Phys., 2007, 45(4), 444–454 CrossRef CAS.
- J. D. Merline, C. P. R. Nair, C. Gouri, R. Sadhana and K. N. Ninan, Eur. Polym. J., 2007, 43, 3629–3637 CrossRef CAS.
- T. S. Wilson, J. P. Bearinger, J. L. Herberg, J. E. Marion III, W. J. Wright, C. L. Evans and D. J. Maitland, J. Appl. Polym. Sci., 2007, 106(1), 540–551 CrossRef CAS.
- S. D'Hollander, G. Van Assche, B. Van Mele and F. Du Prez, Polymer, 2009, 50(19), 4447–4454 CrossRef.
- J. Han, B. Chen, L. Ye, A.-Y. Zhang, J. Zhang and Z.-G. Feng, Front. Mater. Sci. China, 2009, 3(1), 25–32 CrossRef.
- L. Xue, S. Dai and Z. Li, Macromolecules, 2009, 42(4), 964–972 CrossRef CAS.
- M. Ahmad, J. Luo, B. Xu, H. Purnawali, P. J. King, P. R. Chalker, Y. Fu, W. Huang and M. Miraftab, Macromol. Chem. Phys., 2011, 212, 592–602 CrossRef CAS.
- I. M. Pereira and R. L. Oréfice, Macromol. Symp., 2011, 299–300, 190–198 CrossRef.
- A. Petchsuk, W. Klinsukhon, D. Sirikittikul and C. Prahsarn, Polym. Adv. Technol., 2012, 23(8), 1166–1173 CrossRef CAS.
- M. Bothe, F. Emmerling and T. Pretsch, Macromol. Chem. Phys., 2013, 214(23), 2683–2693 CrossRef CAS.
- T. Pretsch, Smart Mater. Struct., 2010, 19(1), 015006 CrossRef.
- M. Bothe and T. Pretsch, Macromol. Chem. Phys., 2012, 213(22), 2378–2385 CrossRef CAS.
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41(12), 2034–2057 CrossRef CAS.
- P. T. Mather, X. Luo and I. A. Rousseau, Annu. Rev. Mater. Res., 2009, 39(1), 445–471 CrossRef CAS.
- T. Pretsch, Polymers, 2010, 2(3), 120–158 CrossRef CAS.
- J. Leng, X. Lan, Y. Liu and S. Du, Prog. Mater. Sci., 2011, 56(7), 1077–1135 CrossRef CAS.
- H. Lu, Soft Matter, 2013, 9(47), 11157–11158 RSC.
- H. Lu and W. M. Huang, Smart Mater. Struct., 2013, 22(10), 105021 CrossRef.
- H. Lu, Y. Yao and L. Lin, Pigm. Resin Technol., 2015, 44(2), 94–100 CrossRef CAS.
- K. K. Julich-Gruner, C. Löwenberg, A. T. Neffe, M. Behl and A. Lendlein, Macromol. Chem. Phys., 2013, 214(5), 527–536 CrossRef CAS.
- G. J. Berg, M. K. McBride, C. Wang and C. N. Bowman, Polymer, 2014, 55(23), 5849–5872 CrossRef CAS.
- T. Xie, K. A. Page and S. A. Eastman, Adv. Funct. Mater., 2011, 21(11), 2057–2066 CrossRef CAS.
- K. Kratz, U. Voigt and A. Lendlein, Adv. Funct. Mater., 2012, 22(14), 3057–3065 CrossRef CAS.
- N. Fritzsche and T. Pretsch, Macromolecules, 2014, 47(17), 5952–5959 CrossRef CAS.
- L. Viry, C. Mercader, P. Miaudet, C. Zakri, A. Derré, A. Kuhn, M. Maugey and P. Poulin, J. Mater. Chem., 2010, 20, 3487–3495 RSC.
- T. Xie, Nature, 2010, 464(10), 267–270 CrossRef CAS PubMed.
- R. Xiao, J. Guo and T. Nguyen, RSC Adv., 2015, 5(1), 416–423 RSC.
- J. Cui, K. Kratz and A. Lendlein, Smart Mater. Struct., 2010, 19(6), 065019 CrossRef.
- J. Li, T. Liu, S. Xia, Y. Pan, Z. Zheng, X. Ding and Y. Peng, J. Mater. Chem., 2011, 21, 12213–12217 RSC.
- C. Samuel, S. Barrau, J.-M. Lefebvre, J.-M. Raquez and P. Dubois, Macromolecules, 2014, 47(19), 6791–6803 CrossRef CAS.
- F. Grillard, C. Zakri, P. Gaillard, A. Korzhenko, W. Néri and P. Poulin, Soft Matter, 2014, 10(44), 8985–8991 RSC.
- J. Hättig, W. Bräuer and T. Pretsch, Kunststoffe, 2013, 1(1), 39–41 Search PubMed.
- T. Pretsch, M. Ecker, M. Schildhauer and M. Maskos, J. Mater. Chem., 2012, 22(16), 7757–7766 RSC.
- N. Fritzsche and T. Pretsch, ASME Conf. Smart Mater., Adapt. Struct. Intell. Syst., Proc., 2012, 1, 81–88 Search PubMed.
- M. Ecker and T. Pretsch, RSC Adv., 2014, 4(1), 286–292 RSC.
- M. Ecker and T. Pretsch, RSC Adv., 2014, 4(87), 46680–46688 RSC.
- DIN EN ISO 527–2:1996, Plastics – Determination of tensile properties.
- K.-H. Nitta and H. Nomura, Polymer, 2014, 55(25), 6614–6622 CrossRef CAS.
- O. Paris, C. Li, S. Siegel, G. Weseloh, F. Emmerling, H. Riesemeier, A. Erko and P. Fratzl, J. Appl. Crystallogr., 2007, 40(3), 466–470 Search PubMed.
- A. P. Hammersley, S. O. Svensson, M. Hanfland, A. N. Fitch and D. Hausermann, High Pressure Res., 1996, 14(4–6), 235–248 CrossRef.
- M. Bothe and T. Pretsch, J. Mater. Chem. A, 2013, 1(46), 14491–14497 CAS.
- M. Bothe, K. Y. Mya, E. M. J. Lin, C. C. Yeo, X. Lu, C. He and T. Pretsch, Soft Matter, 2012, 8(4), 965–972 RSC.
- W. M. Huang, H. B. Lu, Y. Zhao, Z. Ding, C. C. Wang, J. L. Zhang, L. Sun, J. Fu and X. Y. Gao, Mater. Des., 2014, 59(7), 176–192 CrossRef.
- J. W. Maher, R. N. Haward and J. N. Hay, J. Polym. Sci., Polym. Phys. Ed., 1980, 18(11), 2169–2179 CrossRef CAS.
- S. L. Bazhenov and A. S. Kechek'yan, J. Polym. Sci., Part A: Polym. Chem., 2013, 55(6), 404–414 CAS.
- M. A. Guseva, V. A. Gerasin, O. K. Garishin, V. V. Shadrin, O. A. Plekhov and A. Pawlak, Polymer, 2015, 56(1), 416–427 CrossRef CAS.
- J. Gough, Philos. Mag., 1806, 24(93), 39–43 Search PubMed.
- E. Pink, A. Kronthaler and P. Valtingoier, J. Mater. Sci., 1989, 24(1), 183–186 CrossRef CAS.
- F. L. Ji, J. L. Hu, T. C. Li and Y. W. Wong, Polymer, 2007, 48(17), 5133–5145 CrossRef CAS.
- T. Pretsch, I. Jakob and W. Müller, Polym. Degrad. Stab., 2009, 94(1), 61–73 CrossRef CAS.
- T. Pretsch and W. Müller, Polym. Degrad. Stab., 2010, 95(5), 880–888 CrossRef CAS.
- W. Müller and T. Pretsch, Eur. Polym. J., 2010, 46(8), 1745–1758 CrossRef.
- K. Y. Mya, H. B. Gose, T. Pretsch, M. Bothe and C. He, J. Mater. Chem., 2011, 21(13), 4827–4836 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra05492c |
| This journal is © The Royal Society of Chemistry 2015 |