
Enhanced catalytic activity with high thermal stability based on multiple Au cores in the interior of mesoporous Si–Al shells
Abstract
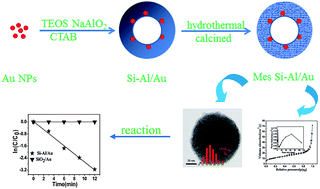
A novel mesoporous Si–Al/Au catalyst with core–shell structure was successfully fabricated by the combination of a sol–gel strategy and calcination process. This method involves the preparation of a gold sol and the capsulation of Si–Al layers. Afterwards, the mesoporous Si–Al/Au catalyst was obtained by calcination at 550 °C to remove the surfactant and other organics. The synthesized samples were characterized by several techniques, including transmission electron microscopy (TEM), energy-dispersive X-ray spectroscopy analysis, X-ray diffraction, field emission scanning electron microscopy (FESEM), N2 adsorption–desorption isotherms and UV-Vis spectra. It was found that this Si–Al/Au core–shell catalyst exhibited high thermal stability and the existence of a mesoporous structure could ensure high permeation and mass transfer rates for species involved in a catalytic reaction. After the calcination, the Au nanoparticles still maintained their small size because of the protective effect of the outside Si–Al layers. Moreover, when the samples were treated by a hydrothermal method, the one core was changed to multiple cores, resulting in the high catalytic activities for the reduction of p-nitrophenol (p-NPh). In our experiment, this prepared catalyst could be easily recycled without a decrease of the catalytic activities in the reaction.
 Please wait while we load your content...
Please wait while we load your content...

