
Amorphous carbon framework stabilized SnO2 porous nanowires as high performance Li-ion battery anode materials†
 a
a
Abstract
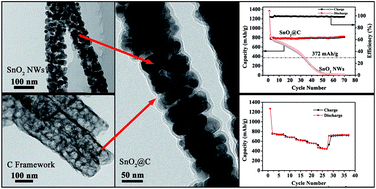
Amorphous carbon framework stabilized SnO2 porous nanowires (SnO2@C nanocomposites) were successfully synthesized through a hydrothermal-calcining method. The in situ formed amorphous carbon framework not only enhances the electron conductivity but also accommodates the volume expansion during the electrochemical cycling process. Benefiting from the structure and amorphous carbon framework, SnO2@C nanocomposites show ultra-excellent cycling performance, rate capability and high coulombic efficiency. At a rate of 782 mA g−1, their reversible capacity is as high as 751 mA h g−1 with no capacity fading after 160 cycles. These experimental findings may provide some insights to further improve the cyclability and rate capability of anode materials, paving the way for the next-generation high performance Li-ion batteries.
 Please wait while we load your content...
Please wait while we load your content...

