
Selective fluorometric detection of F− and Zn(ii) ions by a N, O coordinating sensor and naked eye detection of Cu(ii) ions in mixed-aqueous solution†
Abstract
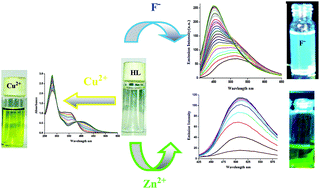
Through click chemistry, salicylaldehyde and fluorene groups have been explored to recognize anions through O–H⋯A– hydrogen-bonding complexation followed by an ESIPT mechanism and cation sensing via CHEF and CHEQ mechanisms for Zn2+ and Cu2+ metal ions respectively. Herein, we demonstrate evidence of the interactions between the O–H bond and F− which were studied by fluorescence spectroscopic, UV-Vis spectroscopic, lifetime spectroscopic, 1H NMR spectroscopic titrations and theoretical treatment. This sensor could simultaneously detect two biologically important metal ions (Cu2+ and Zn2+) through colorimetric methods in mixed aqueous solution. The 2 : 1 binding stoichiometry ratio of the ligand and metal in the complexes was established by UV-Vis, fluorescence, 1H NMR and ESI-MS spectroscopy, and their corresponding association constants, Kassoc observed at 8.13 × 104 and 5.12 × 106 M−1 corresponds to Zn2+ and Cu2+ metal ions in aqueous buffer–CH3OH (2 : 1, v/v) at pH 7.2. In addition, the electronic structures and photo physical properties of the ligand and complexes were calculated by DFT and time-dependent DFT (TDDFT) methods.
 Please wait while we load your content...
Please wait while we load your content...

