
AMPK-mediated crosstalk of heteroglycan-induced reactive species and autophagic cascade in RAW 264.7 cells
K. Sanjana P. Devi†
,
Joyjyoti Das†,
Kalpana Kumari†,
Prashant Singh,
Birendra Behera and
T. K. Maiti*
Department of Biotechnology, Indian Institute of Technology Kharagpur, Kharagpur 721302, West Bengal, India. E-mail: tkmaiti@hijli.iitkgp.ernet.in; Fax: +91 03222 278707; Tel: +91 3222 283766
First published on 23rd April 2015
Abstract
Natural polysaccharides are currently being employed for macrophage activation due to their minimal cytotoxicity and side effects. Reactive species like nitric oxide (NO) and reactive oxygen species (ROS), induced by a novel immunostimulatory heteroglycan were found to regulate the autophagic machinery in RAW 264.7 cells via a feedback mechanism, through AMPK. Heteroglycan-induced autophagy enhanced NO generation while itself being negatively regulated by NO. Concomitantly, intracellular ROS triggered the autophagic machinery which in turn subdued ROS levels indicating a homeostatic mechanism for regulated immune function. This study may help in better understanding of regulated immunostimulation by natural polysaccharides.
1. Introduction
In a healthy immune system, macrophages perform a variety of complex microbicidal functions, including surveillance, chemotaxis, phagocytosis and destruction of targeted organisms.1 Though phagocytes defend against a host of pathogenic microbes like fungi, bacteria and virus infected cells, the emergence of new pathogens is a serious problem, and must be dealt with meticulously by physicians. Thus, the development of novel therapeutics, which could nonspecifically augment the innate immune response, represents an ideal strategy for addressing current worldwide concerns of how to combat classical and emerging infectious agents. Plant polysaccharides have been found to be ideal candidates for therapeutics with immunomodulatory functions since they are relatively non-toxic and do not cause significant side effects. In particular, these compounds have been shown to increase macrophage cytotoxic activity against tumor cells and microorganisms, activate phagocytic activity, increase reactive oxygen species (ROS) and nitric oxide (NO) production, and enhance secretion of cytokines and chemokines such as tumor necrosis factor (TNF-α), interleukin (IL)-1β, IL-6, IL-8, IL-12, IFN-γ and IFN-β2.2 Activation of macrophages is essential for the host defense system and accumulating evidence suggests that autophagy, a cellular homeostatic mechanism is an important component in macrophage activity and hence immune functioning.3,4 Prolonged activation of macrophages, however, becomes detrimental and contributes to the progression of chronic inflammatory diseases with abnormal cell death. Therefore, controlling the over-activation of macrophages may be a new therapeutic strategy for various inflammatory diseases.5 With this intent, we studied the probable crosstalk between heteroglycan-induced reactive species and autophagy in RAW 264.7 macrophages induced by a novel heteroglycan isolated from mycelia of an edible mushroom, Pleurotus ostreatus.ROS and nitric oxide are known to modulate autophagy in various cell types.6,7 AMPK is a major mediator of indirect activation of autophagy by ROS, as it is sensitive to oxidative stress induced by H2O2 and gets phosphorylated at Thr172 of its α1 catalytic subunit.8 AMPK is also known to be involved in regulation of autophagy by reactive nitrogen species.9 Therefore studying the role of this molecule in macrophage autophagy and immune function could be instrumental in finding the key molecular targets for controlling activation of macrophages. In our study, we used a heteroglycan isolated from the mycelia of Pleurotus ostreatus which had been completely characterized in previous reports.10,11 The monomeric repeating unit of the polysaccharide molecule is:
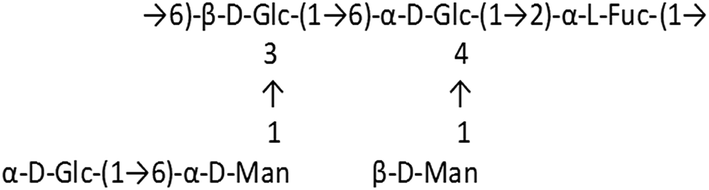
The isolated heteroglycan was found to possess immunomodulatory properties and can also lead to tumor regression.10 Fungal cell walls have highly conserved structural components composed of polysaccharides that potently trigger innate immune responses.12 Several polysaccharide binding receptors such as Dectin-1 and TLR2 have also been related to physiological processes such as autophagy.13,14
We observed though our experiments that the heteroglycan induces the generation of reactive oxygen species and nitric oxide which regulate autophagy positively and negatively respectively, through AMPK. Heteroglycan-induced autophagy aids in NO production and reduces ROS levels in cell thereby indicating that it might be playing a crucial role in immune functioning through sustained cellular homeostasis. To the best of our knowledge, this is the first study on the interplay between autophagy and reactive species induced by a natural heteroglycan and it may have great implications towards an in-depth understanding of polysaccharide-triggered immunomodulation.
2. Materials & methods
2.1. Isolation of heteroglycan
Heteroglycan was isolated from the mycelia of Pleurotus ostreatus by an established protocol which has already been reported earlier.10 Extraction was based on an alkali treatment method, followed by ethanol precipitation and anion exchange chromatography of the precipitated polysaccharides. Samples were later lyophilized and preserved at −20 °C until further use.2.2. Cell line maintenance
RAW 264.7 cells (obtained from NCCS, Pune) were maintained in DMEM with 10% FBS and antibiotic supplementation in a 5% CO2 incubator at 37 °C.2.3. Acridine orange staining to quantify acidic vacuoles (AVOs) formation
RAW 264.7 cells were seeded at a concentration of 2 × 106 cells per ml in six well plates and treated with 50, 100, 150 and 200 μg ml−1 of heteroglycan with or without 1 h pretreatment with the autophagy inhibitor 3-MA (3-methyl adenine). 3-MA is known as an early stage inhibitor of autophagy. It acts by inhibiting the class III PtdIns3K.15,16 It is used as 1 h pre-treatment, at a concentration of 10 mM. 3-MA was used at all instances where the effect of autophagy on a particular phenomenon was needed to be studied. Cells treated with heteroglycan (with or without 3-MA pre-treatment) were then incubated for a period of 16 h, followed by scraping and staining with acridine orange (5 μg ml−1) for 15 min to visualize acidic compartments under fluorescence microscope, as a preliminary indication of autophagy. The presence of acidic vacuoles (AVOs) was also quantified by flow cytometry with acridine orange staining using a FACS Calibur flow cytometer (Becton-Dickinson, USA).2.4. Autophagy detection by confocal microscopy of EGFP-LC3
Autophagy was analysed by transfecting RAW 264.7 cells with the ptfLC3 plasmid which was received as a gift from Tamotsu Yoshimori (Addgene plasmid # 21074). The GFP-LC3 fusion protein is under the expression of a CMV promoter. It's a constitutive mammalian promoter.17 Hence, the level of expression is not controlled by any external factors. The plasmid (Addgene plasmid ptfLC3, plasmid # 21074) containing the genes, is well characterized and used for detection of autophagy. Lipofectamine® 2000 (Life Technologies) was used for transfection. 3 × 105 cells per ml were plated and allowed to adhere overnight and transfected according to a protocol described elsewhere.18 DNA to Lipofectamine ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 was found to yield highest transfection efficiency. Autophagosomes (puncta) were detected by laser excitation at 488 nm to monitor EGFP-LC3 puncta formation, by Olympus FV-1000 Confocal microscope. For puncta counting, Z-sections of images from 10 different fields of view were taken per sample through 63× oil immersion objective on an Olympus IX81 Confocal microscope. The number of GFP-LC3 puncta in each cell was counted visually by an observer blinded to the experimental conditions and averaged over 10 frames.19–21 At least 50 cells per condition were taken into consideration.
:2 was found to yield highest transfection efficiency. Autophagosomes (puncta) were detected by laser excitation at 488 nm to monitor EGFP-LC3 puncta formation, by Olympus FV-1000 Confocal microscope. For puncta counting, Z-sections of images from 10 different fields of view were taken per sample through 63× oil immersion objective on an Olympus IX81 Confocal microscope. The number of GFP-LC3 puncta in each cell was counted visually by an observer blinded to the experimental conditions and averaged over 10 frames.19–21 At least 50 cells per condition were taken into consideration.
ptfLC3 transfected RAW 264.7 cells were seeded at a density of 3 × 105 cells per ml. Heteroglycan was used at various concentrations i.e., 100 μg ml−1, 150 μg ml−1 and 200 μg ml−1. To determine the change in autophagic flux, Bafilomycin A1 was used. Bafilomycin A1 (baf) is a V-ATPase inhibitor that causes an increase in lysosomal/vacuolar pH, and ultimately blocks fusion of autophagosomes with the vacuole.22,23 It is used as 1 h pre-treatment at a concentration of 100 nM. For assessment of autophagic flux in cells, the maturation of autophagosomes to autophagolysosomes was inhibited by inactivating lysosomal degradation. Autophagosomes in the cells with and without heteroglycan treatment for 16 h were therefore quantified to determine the autophagic flux.
2.5. Confocal microscopy to study effect of NO and ROS on heteroglycan induced autophagy
Cells transiently transfected with EGFP-LC3 were pre-treated with different inhibitors like N-acetyl cysteine (NAC, 1 h pre-treatment, 5 mM), N6-(1-iminoethyl)-L-lysine (L-NIL, 1 h pre-treatment, 1 μM) and Bafilomycin A1 (Baf, 1 h pre-treatment, 100 nM) and then treated with heteroglycan (150 μg ml−1) for 16 h. NAC is a reactive oxygen species inhibitor24,25 and is used as 1 h pre-treatment at a concentration of 10 mM. Bafilomycin A1 (baf) prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes.24,26 L-NIL (L-N6-(1-iminoethyl) lysine dihydrochloride) on the other hand is a selective inhibitor of inducible nitric oxide synthase (iNOS).26,27 L-NIL is a standard inhibitor of NO and was used in our experiments to assess the effect of NO on autophagy. The cells which were treated with heteroglycan and various inhibitors were then observed under confocal microscope for puncta (autophagic vacuole) formation.2.6. Nitric oxide assay
Nitric oxide (NO) production was measured to determine the in vitro activation of stimulated cells. RAW 264.7 cells were adjusted to a density of 1 × 106 cells per ml of which 180 μl was plated in a 96 well plate. Cells were allowed to adhere for few hours and then positive control (LPS) and heteroglycan (150 μg ml−1, as found from the results of Section 2.3) with and without autophagy inhibitor viz. 3-methyl adenine (initial step inhibitor, 10 mM 1 h pre-treatment) was added to the respective wells. Treated cells were then incubated for 24 h at 37 °C in a 5% CO2 incubator and NO production assayed by estimating the nitrite concentration in the cell supernatant with Griess reagent. Equal volumes of isolated supernatants and Griess reagent were mixed and incubated at room temperature for 10 min, following which, absorbance was recorded at 540 nm.282.7. Detection of ROS by DCFDA staining
RAW 264.7 cells (2 × 106 cells per ml) were treated with 150 μg ml−1 of heteroglycan in the presence/absence of 10 mM 3-MA (3-methyl adenine, 1 h pre-treatment) for 16 h in a CO2 incubator. LPS was taken as the positive control as it stimulates production of reactive oxygen species. Thereafter, cells were stained with 20 mM DCFDA (2′-7′-dichlorodihydrofluorescein diacetate) for 15 min, washed with PBS and analysed using Olympus FV-1000 Confocal microscope. The images were analysed using ImageJ version 1.48. The regions of interest (ROI) in image were selected by using freehand drawing/selection tools (at least 5 ROIs/image were taken into consideration). The fluorescence intensity was determined for all ROIs in each image and mean fluorescence intensity (MFI) were calculated by taking average of measured fluorescence intensity of ROIs in the image.2.8. Western blotting
RAW 264.7 cells were treated with 150 μg ml−1 of heteroglycan for 16 h after pre-treatment with or without Bafilomycin A1 or Compound C (an AMPK inhibitor). Compound C or Dorsomorphin acts as a potent, selective, reversible, and ATP-competitive inhibitor of AMPK (AMP-activated protein kinase). It was used at a working concentration of 10 μM (1 h pre-treatment). Compound C is often used as an AMPK inhibitor in various studies related to AMPK signalling.29,30 In another set of experiments, RAW264.7 cells were pre-treated with different inhibitors like N-acetyl cysteine (NAC, 1 h pre-treatment, 5 mM), N6-(1-iminoethyl)-L-lysine (L-NIL, 1 h pre-treatment, 1 μM) Compound C (1 h pre-treatment, 10 μM) and Bafilomycin A1 (Baf, 1 h pre-treatment, 100 nM), which was then followed by heteroglycan treatment for 16 h to understand the role of AMPK in the crosstalk of heteroglycan-induced reactive species and autophagy. Since, we were interested to examine the effect of reactive oxygen species and NO on autophagy and AMPK regulation, it was essential to inhibit ROS and NO using known standard inhibitors.The protein lysates collected from control and heteroglycan treated cells (with and without inhibitor pre-treatment) using cell lysis buffer were resolved in a 15% SDS-PAGE and transferred in a polyvinylidene fluoride (PVDF) membrane. Non-specific protein blocking of the membrane was done for 1 h at room temperature with 2% bovine serum albumin followed by incubation with primary antibodies specific for LC3B, AMPK, p-AMPK and β-actin (2–4 h at room temperature). All antibodies were purchased from Cell Signaling Technology, USA. Blots were then washed and further incubated with suitable HRP-conjugated secondary antibodies at room temperature for 1 h. Membranes were developed with the signal producing Chemiluminescent Western Blotting kit (Sigma, USA) and exposed to an X-ray film (Eastman Kodak) for image development. The immunoblots were analysed using ImageJ software. The intensities of the bands were estimated by the percentage area of peak obtained from each band after subjecting the blot images for software analysis.
2.9. Statistical analysis
All statistical analyses were performed in MS Excel and stated as mean ± standard deviation. Significance of the data was examined by t-test for unpaired observations with *P-value <0.05.3. Results
3.1. Heteroglycan induces acidic vacuole formation due to increased autophagy
Acridine orange is a fluorescent dye which can intercalate in nucleic acids (and emit green fluorescence) as well as enter in acidic vacuoles such as lysosomes wherein they become protonated and fluoresce orange light. In the present work, epifluorescent microscopic imaging of acridine orange stained RAW 264.7 cells revealed that heteroglycan treatment at different concentrations significantly increased the formation of acidic vacuoles (AVOs) (Fig. 1A) in comparison to control indicating the increasing delivery of autophagosomes to lysosomes. Several time points of study upto 24 h were chosen with intervals of 4 h and maximum AVOs were found at 16 h (data not shown here). It was confirmed though dot plot analysis of FACS data that maximum AVOs (indicated by increased FL3-H) were produced at a concentration of 150 μg ml−1 heteroglycan treatment (Fig. 1B). A further increase in the dosage (200 μg ml−1 heteroglycan treatment) decreased the percentage of AVOs in RAW 264.7 cells. Upon treatment with an early-autophagy inhibitor 3-methyladenine, FL3-H (red) intensity reduced significantly confirming that heteroglycan induces autophagy in RAW 264.7 cells (Fig. 1C). Henceforth, the concentration of 150 μg ml−1 at which maximum AVOs were noted for heteroglycan was used consistently in further experiments.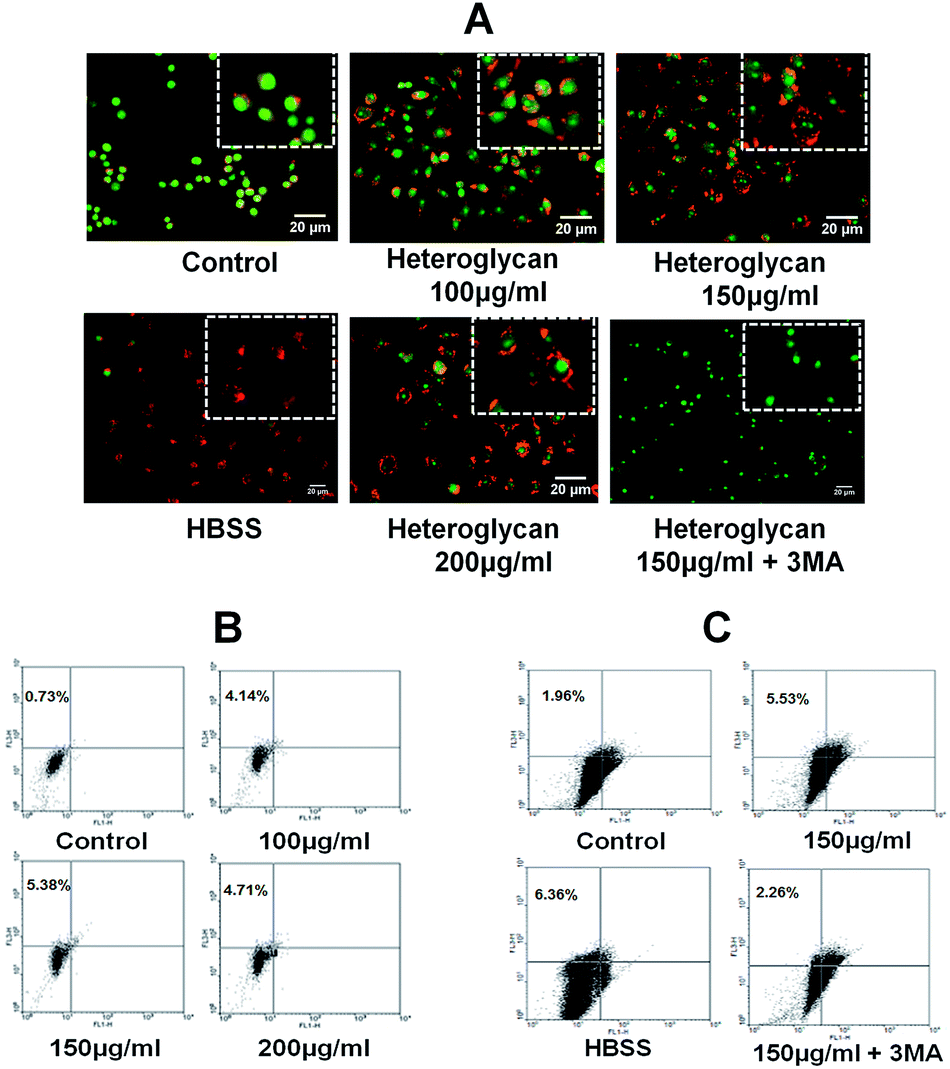 | ||
| Fig. 1 Heteroglycan induces autophagy. (A) Fluorescence microscopic images of RAW 264.7 cells stained with acridine orange (5 μg ml−1) after treatment with different heteroglycan concentrations (with scale bar 20 μm) for 16 h. (B) Dot plot analysis of different doses of heteroglycan-treated cells with acridine orange staining, obtained though flow cytometry. Upper left region of dot plots displays the percentage of AVOs (FL3-H). (C) Dot plot analysis of heteroglycan-treated cells with or without autophagy inhibitor, 10 mM 3-methyladenine. Hanks' Balanced Salt solution (HBSS)-treated cells were used as positive control by inducing autophagy through serum starvation for 6 hours. | ||
3.2. Heteroglycan-induced autophagy aids in NO production and downregulates ROS
The heteroglycan was found to increase nitric oxide production in RAW 264.7 cells. However, when autophagy was inhibited by the addition of 3-MA during heteroglycan treatment, nitric oxide production decreased significantly by 63% (Fig. 2A) in comparison to cells which were subjected to heteroglycan treatment without 3-MA inhibition. ROS assay by DCFDA staining on the other hand indicated that heteroglycan induces ROS generation in RAW 264.7 cells and it was found to be down-regulated by autophagy (Fig. 2B(i) and B(ii)). Autophagy inhibition by 3-MA increased ROS production in RAW 264.7 cells treated with heteroglycan by 52% compared to cells without autophagy inhibition.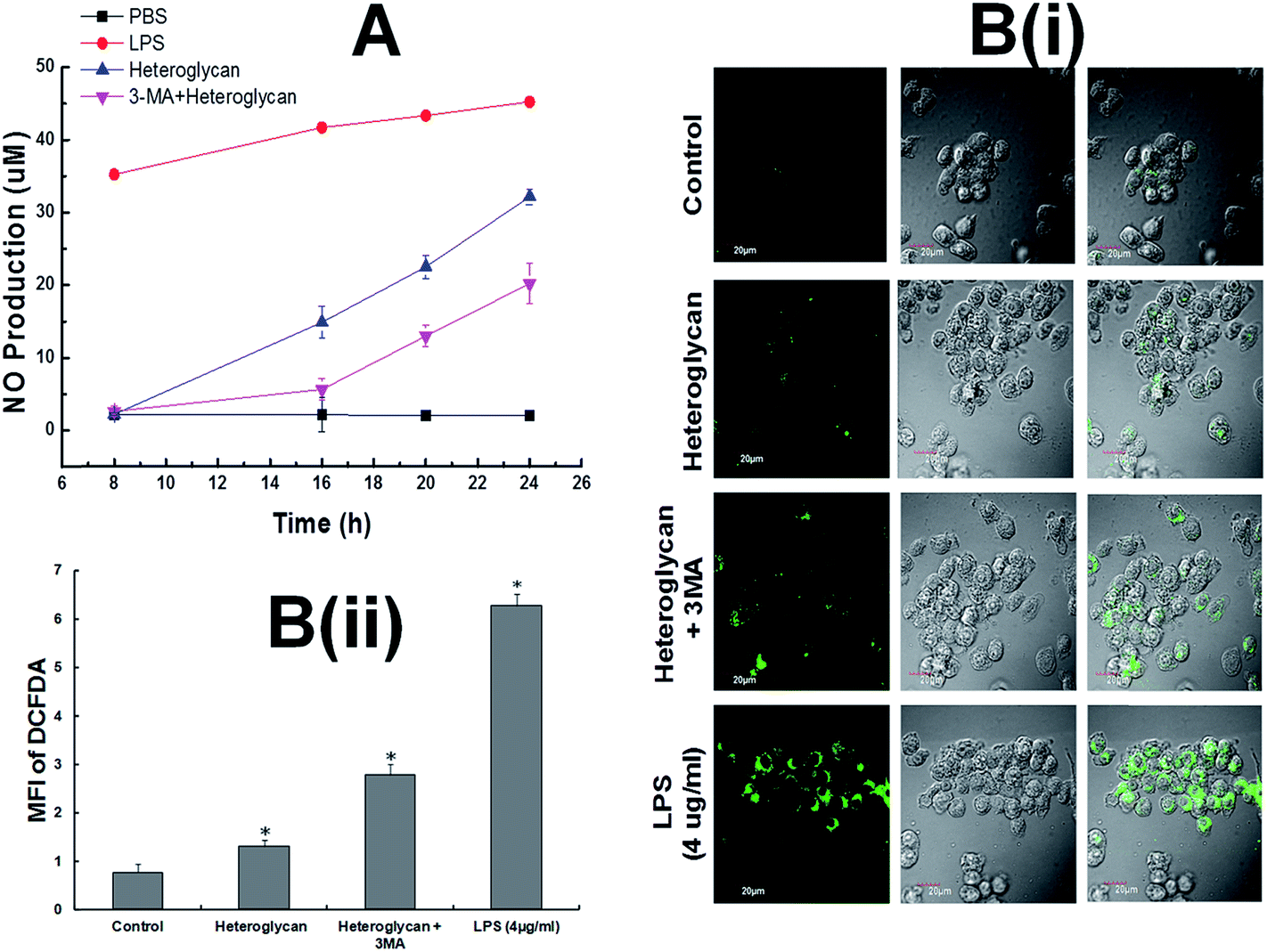 | ||
| Fig. 2 Heteroglycan produces NO and ROS. (A) NO production in RAW 264.7 cells by heteroglycan with and without inhibition of autophagy. (B(i)) Confocal microscopy images of DCFDA-stained heteroglycan-treated RAW 264.7 cells with or without autophagy inhibition (with scale bar = 20 μm) for 16 h. (B(ii)) Comparative analysis of mean fluorescence intensity of DCFDA from different samples. Lipopolysaccharide was kept as a positive control for both NO and ROS assay. All values are represented as mean ± SD (n = 6) and significance was determined using t-test statistics with *P < 0.05. | ||
3.3. ROS enhances autophagy while NO regulates it through a negative feedback loop
Confocal microscopy of EGFP-LC3 puncta showed that when nitric oxide was inhibited, autophagic flux increased significantly suggesting that the predominance of ROS caused by heteroglycan might have positively upregulated autophagosome formation (Fig. 3). Contrastingly, when ROS was inhibited with NAC, the number of autophagosomes decreased, exhibiting a more diffuse form of EGFP-LC3, which indicated that the predominance of nitric oxide in the cell might have reduced the autophagic flux significantly (Fig. 3). | ||
| Fig. 3 NO downregulates autophagy while ROS upregulates it. Z-scanning confocal microscopy of EGFP-LC3 autophagic puncta in transfected RAW 264.7 cells treated with heteroglycan and inhibitors of NO (L-NIL) and ROS (NAC) in presence or absence of Bafilomycin A1 to measure autophagic flux, after 16 h of treatment. At least 10 frames per sample were taken into consideration. All values are represented as mean ± SD (n = 6) and significance was determined using t-test statistics with *P < 0.05. | ||
3.4. Heteroglycan-induced ROS and NO regulate autophagy though the intervention of AMPK
LC3 is the most well known marker of autophagy.31,32 LC3-I, a microtubule-associated protein 1 (MAP1) light chain 3 is normally present in the cytosol and conjugates with phosphatidylethanolamine on autophagy activation to form LC3-II which then gets localized in the membrane of the autophagosomes. Jang et al., 2013 have demonstrated that autophagic flux can be determined by measuring the conversion of LC3-I to LC3-II which finally gets incorporated in the autophagosomes.33 Upon heteroglycan stimulation on RAW 264.7 cell line for 16 h, increase in 50% of LC3-II/LC3-I ratio was observed with respect to control cells when the end steps of autophagy were impaired by lysosomal inactivation. Furthermore, on inhibition of p-AMPK by Compound C, it was noted that cells which were subjected to heteroglycan and Bafilomycin treatment showed extremely low levels of LC3-II/LC3-I ratio (74%) than cells with only Bafilomycin treatment (Fig. 4A). Correspondingly, AMPK phosphorylation to p-AMPK increased by 20% on heteroglycan treatment. In presence of heteroglycan-induced ROS with NO inhibition (heteroglycan + L-NIL) phosphorylation was found to be 40% increased. On the contrary, in case of heteroglycan-induced NO predominance with ROS inhibition (heteroglycan + NAC), the level of phosphorylation declines by 30% in comparison to that in heteroglycan-treated cells (Fig. 4B), clearly indicating that AMPK phosphorylation is influenced by heteroglycan-induced reactive species.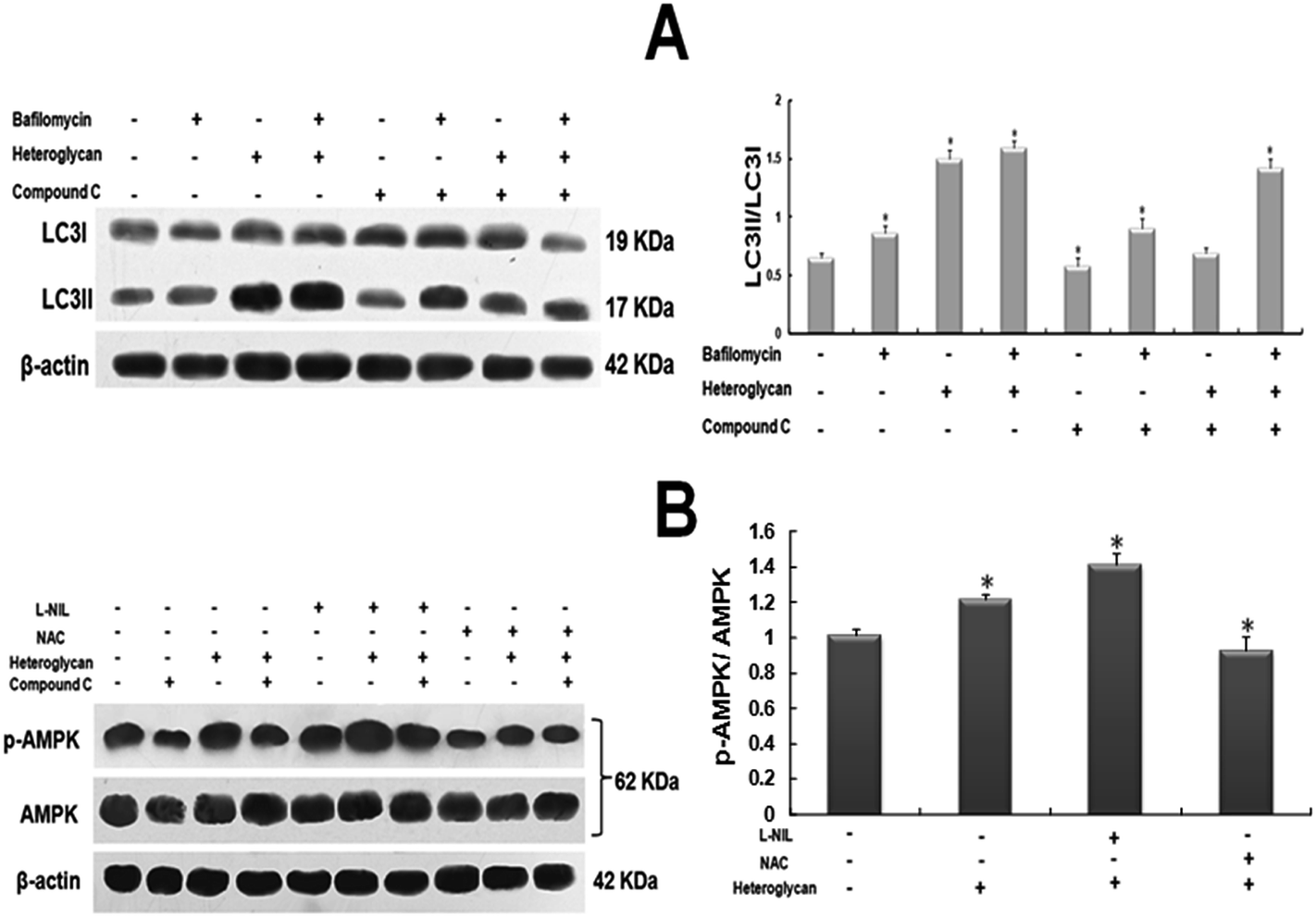 | ||
| Fig. 4 Heteroglycan induced ROS and NO regulate autophagy through AMPK. (A) Western blot of LC3 for heteroglycan-treated cells with or without inhibitor of AMPK (Compound C), in presence or absence of Bafilomycin A1, after 16 h of treatment. Comparative analysis shows LC3-II/LC3-I ratios (indicative of autophagic flux) of various samples with respect to control. (B) Western blot of p-AMPK and AMPK for cells treated with heteroglycan and inhibitors of NO (L-NIL) and ROS (NAC), in presence or absence of Bafilomycin A1, after 16 h of treatment. Comparative analysis shows p-AMPK/AMPK ratios of various samples with respect to control. All values are represented as mean ± SD (n = 6) and significance was determined using t-test statistics with *P < 0.05. | ||
4. Discussion
Reactive species such as NO and ROS are pivotal signaling molecules which elicit a multitude of physiological processes through diverse mechanisms. These molecules are highly essential for activating macrophages for host defense. However, an imbalance in their generation inside the mitochondria disrupts the cellular redox homeostasis and leads to the onset of nitrosative or oxidative stress which has been etiologically linked to various pathological conditions.7 Here, we investigate the role of heteroglycan in inducing autophagy (a cellular homeostatic mechanism) in immune cells and also correlate its inherent potential of generating NO and ROS with autophagic regulation. Despite many findings in the past, the interplay between nitric oxide and mitochondrial ROS levels and autophagy still remains enigmatic due to their complex mode of action. Furthermore, such studies have not been reported in immune cells. In our study, we used a well-characterized heteroglycan isolated from the mycelia of an edible mushroom, P. ostreatus and confirmed the induction of autophagy in RAW 264.7 cells by the increase (or decrease on 3-MA pretreatment) in the formation of acidic vacuoles (AVOs) and change in autophagic flux over 16 h. We noted that heteroglycan-induced autophagy augmented nitric oxide production while reducing ROS levels in RAW 264.7 cells. This phenomenon may be linked with ROS accumulation in autophagy-deficient cells. It is known that autophagy maintains the normal mitochondrial turnover by a process known as mitophagy and inhibition of mitophagy abnormally increases the intracellular ROS levels generated from defective mitochondria.34 High ROS can cause oxidative modification of nitric oxide synthase (NOS) and disrupts the electron transfer within the enzyme thereby impeding NO formation from L-arginine.7,35On the other hand, we found that ROS and NO show antagonistic effects on heteroglycan-induced autophagy indicating that reactive nitrogen and oxygen species also regulate the “self eating” process in macrophage cells. Reactive oxygen species (ROS) was found to positively regulate autophagy in the present case. An earlier report with similar finding claimed that in NIH-3T3 cells, ROS induces autophagy through AMPK which senses oxidative stress and gets phosphorylated at its catalytic site (Thr 172) to further inhibit mTORC1 and increase autophagosome formation.8 This suggests that there exist many other mechanisms which impede the action of mTOR to maintain cellular homeostasis and induce autophagy under conditions of oxidative stress. Contrary to the effect of ROS, nitric oxide (NO) production by heteroglycan was found to exhibit a negative regulation on autophagic processes in RAW 264.7 cells. The inhibitory effect of NO on autophagy involves a complex mechanism and is associated with a number of variable factors which are governed at the protein level. It is reported that nitric oxide impairs autophagy by two independent mechanisms in mammalian cells: (1) NO reduces JNK1 phosphorylation and abrogates the phosphorylation of anti-autophagy protein Bcl2 leading to a robust Bcl2-Beclin1 association, which disrupts hVps34/Beclin1 complex formation and thereby inhibits autophagy in HeLa cells;6 (2) NO also causes S-nitrosylation of IKKβ and hinders AMPK phosphorylation, which, in turn, leads to reduction of autophagy.6,36,37 The impact of NO accumulation on autophagy inhibition has been demonstrated to be due to the overexpression of iNOS, a Ca2+ independent enzyme which catalyzes nitric oxide production on stimulation.6 In the context of heteroglycan-induced autophagy, our dataset also suggests that beyond a threshold level, NO produced on heteroglycan treatment was able to cause reduction in the formation of autophagosomes in RAW 264.7 cells (Fig. 3). A schematic representation of the pathways reported in this study through which reactive nitrogen and oxygen species regulate autophagy in RAW 264.7 cells is provided in Fig. 5.
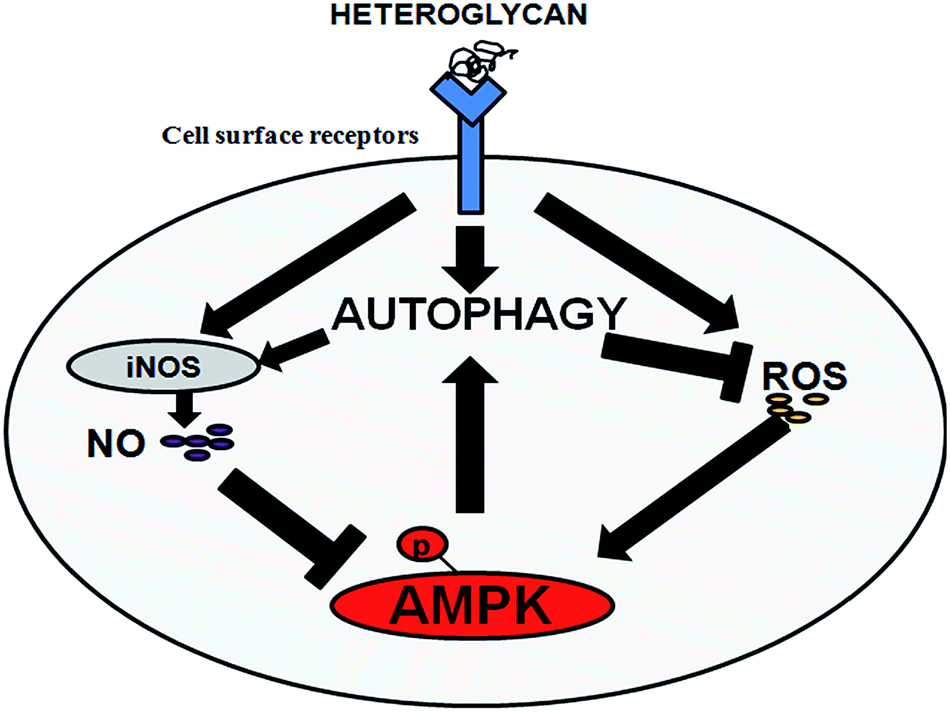 | ||
| Fig. 5 Interplay: schematic representation of the crosstalk between heteroglycan-induced reactive species and autophagy in RAW 264.7 cells explored in the present study. | ||
Reactive species and autophagy seem to play an interdependent role in immune functioning of macrophages and an imbalance in either of them might hamper macrophage immunomodulation. As most of the earlier reports insinuated the involvement of AMPK in the modulation of autophagy by reactive nitrogen and oxygen species, we found it worthwhile to investigate the role of this molecule in this present study. Western blotting results in the present study were clearly suggestive of the fact that AMPK acts as a central molecule regulating heteroglycan-induced reactive species-mediated autophagy in RAW 264.7 cells. This finding thereby provides an initial base to understand the relationship between these processes in macrophages. A deeper insight of the underlying interconnecting pathways may be seminal in the discovery of adjuvant therapeutics for controlled immunostimulation with natural polysaccharides. We hope this report directs researchers to delve into the detailed molecular mechanisms which may thereby aid in designing strategies to modulate autophagy for the treatment of cancer through immune cells.
Conflict of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the Indian Institute of Technology Kharagpur, India for funding the research and the Central Research Facility (CRF) of IIT Kharagpur for technical support.References
- I. A. Schepetkin and M. T. Quinn, Int. Immunopharmacol., 2006, 6, 317 CrossRef CAS PubMed.
- I. A. Schepetkin, G. Xie, L. N. Kirpotina, R. A. Klein, M. A. Jutila and M. T. Quinn, Int. Immunopharmacol., 2008, 8, 1455 CrossRef CAS PubMed.
- B. Levine, Nature, 2007, 446, 745 CrossRef CAS PubMed.
- N. Mizushima, B. Levine, A. M. Cuervo and D. J. Klionsky, Nature, 2008, 451, 1069 CrossRef CAS PubMed.
- H. J. Park, J. H. Yim, H. K. Lee and S. Pyo, FASEB J., 2013, 27, 888 Search PubMed.
- S. Sarkar, V. I. Korolchuk, M. Renna, S. Imarisio, A. Fleming, A. Williams, M. Garcia-Arencibia, C. Rose, S. Luo, B. R. Underwood, G. Kroemer, C. J. O'Kane and D. C. Rubinsztein, Mol. Cell, 2011, 43, 19 CrossRef CAS PubMed.
- J. Lee, S. Giordano and J. Zhang, Biochem. J., 2012, 441, 523 CrossRef CAS PubMed.
- S. L. Choi, S. J. Kim, K. T. Kim, J. Mu, M. J. Birnbaum, S. S. Kim and J. Ha, Biochem. Biophys. Res. Commun., 2001, 287, 92 CrossRef CAS PubMed.
- K. Inoki, T. Zhu and K. L. Guan, Nat. Cell Biol., 2003, 115, 577 CAS.
- K. S. P. Devi, B. Roy, P. Patra, B. Sahoo, S. S. Islam and T. K. Maiti, Carbohydr. Polym., 2013, 94, 857 CrossRef CAS PubMed.
- S. Patra, P. Patra, K. K. Maity, S. Mandal, S. K. Bhunia, B. Dey, K. S. P. Devi, S. Khatua, K. Acharya, T. K. Maiti and S. S. Islam, Carbohydr. Res., 2013, 368, 16 CrossRef CAS PubMed.
- F. McCann, E. Carmona, V. Puri, R. E. Pagano and A. H. Limper, Infect. Immun., 2005, 73, 6340 CrossRef CAS PubMed.
- M. A. Sanjuan, C. P. Dillon, S. W. G. Tait, S. Moshiach, F. Dorset, S. Connell, M. Komatsu, K. Tanaka, J. L. Cleveland, S. Withoff and D. R. Green, Nature, 2007, 450, 1253 CrossRef CAS PubMed.
- J. Ma, C. Becker, C. A. Lowell and D. M. Underhill, J. Biol. Chem., 2012, 287, 34149 CrossRef CAS PubMed.
- E. F. C. Blommaart, U. Krause, J. P. M. Schellens, H. Vreeling-Sindelárová and A. J. Meijer, Eur. J. Biochem., 2004, 243, 240 Search PubMed.
- P. O. Seglen and P. B. Gordon, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 1889 CrossRef CAS.
- S. Kimura, T. Noda and T. Yoshimori, Autophagy, 2007, 3, 452 CrossRef CAS.
- B. Dalby, S. Cates, A. Harris, E. C. Ohki, M. L. Tilkins, P. J. Price and V. C. Ciccarone, Methods, 2004, 33, 95 CrossRef CAS PubMed.
- X. Li-Harms, S. Milasta, J. Lynch, C. Wright, A. Joshi, R. Iyengar, G. Neale, X. Wang, Y. D. Wang, T. A. Prolla, J. E. Thompson, J. T. Opferman, D. R. Green, J. Schuetz and M. Kundu, Blood, 2015, 125, 162 CrossRef CAS PubMed.
- S. Tang, C. Chen, Z. Klase, L. Zane and K. T. Jeang, J. Virol., 2013, 87, 1699 CrossRef CAS PubMed.
- N. Mizushima, T. Yoshimori and B. Levine, Cell, 2010, 140, 313 CrossRef CAS PubMed.
- A. Yamamoto, Y. Tagawa, T. Yoshimori, Y. Moriyama, R. Masaki and Y. Tashiro, Cell Struct. Funct., 1998, 23, 33 CrossRef CAS.
- J. J. Shacka, B. J. Klocke, M. Shibata, Y. Uchiyama, G. Datta, R. E. Schmidt and K. A. Roth, Mol. Pharmacol., 2006, 69, 1125 CrossRef CAS PubMed.
- M. Zafarullah, W. Q. Li, J. Sylvester and M. Ahmad, Cell. Mol. Life Sci., 2003, 60, 6 CrossRef CAS.
- I. A. Cotgreave, Adv. Pharmacol., 1997, 38, 205 CAS.
- W. M. Moore, R. K. Webber, G. M. Jerome, F. S. Tjoeng, T. P. Misko and M. G. Currie, J. Med. Chem., 1994, 37, 3886 CrossRef CAS.
- S. Thoma-Uszynski, S. Stenger, O. Takeuch, M. T. Ochoa, M. Engele, P. A. Sieling, P. F. Barnes, M. Röllinghoff, P. L. Bölcskei, M. Wagner, S. Akira, M. V. Norgard, J. T. Belisle, P. J. Godowski, B. R. Bloom and R. L. Modlin, Science, 2001, 291, 1544 CrossRef CAS PubMed.
- I. Sarangi, D. Ghosh, S. K. Bhutia, S. K. Mallick and T. K. Maiti, Int. Immunopharmacol., 2006, 6, 1287 CrossRef CAS PubMed.
- S. K. Bhutia, T. P. Kegelman and S. K. Das, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22243 CrossRef CAS PubMed.
- J. Jin, T. D. Mullen and Q. Hou, J. Lipid Res., 2009, 50, 2389 CrossRef CAS PubMed.
- X. H. Liang, S. Jackson, M. Seasman, K. Brown, B. Kempkes and H. Hibshoosh, Nature, 1999, 402, 672 CrossRef CAS PubMed.
- Y. Kabeya, N. Mizushima, T. Ueno, A. Yamamoto, T. Kirisako, T. Noda, E. Kominami, Y. Ohsumi and T. Yoshimori, EMBO J., 2000, 19, 5720 CrossRef CAS PubMed.
- B. G. Jang, B. Y. Choi, J. H. Kim, M. J. Kim, M. Sohn and S. W. Suh, PLoS One, 2013, 8, e76466 CAS.
- M. D. Williams, H. V. Remmen, C. C. Conrad, T. T. Huang, J. Charles and A. Richardson, J. Biol. Chem., 1998, 273, 28510 CrossRef CAS PubMed.
- U. Förstermann, Biol. Chem., 2006, 387, 1521 CrossRef PubMed.
- Y. Kondo, T. Kanzawa, R. Sawaya and S. Kondo, Nat. Rev. Cancer, 2005, 5, 726 CrossRef CAS PubMed.
- D. A. Guertin and D. M. Sabatini, Sci. Signaling, 2009, 2, pe24 CrossRef PubMed.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2015 |