
Radical copolymerisation of chlorotrifluoroethylene with isobutyl vinyl ether initiated by the persistent perfluoro-3-ethyl-2,4-dimethyl-3-pentyl radical†
Gerard Putsa,
Gérald Lopezb,
Taizo Onoc,
Philip Crousea and
Bruno Ameduri*b
aDepartment of Chemical Engineering, University of Pretoria, Pretoria 0002, South Africa
bInstitut Charles Gerhardt, Ingénierie et Architectures Macromoléculaires, UMR CNRS 5253, Ecole Nationale Supérieure de Chimie de Montpellier, 8 Rue de l'Ecole Normale, 34296 Montpellier, France. E-mail: bruno.ameduri@enscm.fr
cNational Institute of Advanced Industrial Science and Technology, Research Institute of Instrumentation Frontier, 2266-98, Anagahora, Shimoshidami, Moriyama, Nagoya, Aichi 463-8560, Japan
First published on 29th April 2015
Abstract
Results of the radical copolymerisation of chlorotrifluoroethylene (CTFE) with isobutyl vinyl ether (iBuVE) initiated by ˙CF3 radicals generated by β-scission of perfluoro-3-ethyl-2,4-dimethyl-3-pentyl radical (PPFR) at 90 °C in a batch reactor are reported. 19F NMR spectroscopy enabled the assessment of the molecular weights of the poly(CTFE-alt-iBuVE) copolymer by end-group analysis. It was found that, at low initiator concentrations (≤10 mol%), the ˙CF3 radicals preferably attack the vinyl ether monomer to initiate chain propagation and produce alternating poly(CTFE-alt-iBuVE) copolymers. At initiator ratios of 20 mol%, 19F NMR signals in the CF3 region other than the expected CH2–CF3 are observed and are attributed to ˙CF3 addition patterns due to kinetic effects brought on by monomer solubility. The molecular weights for the copolymer produced from 1%, 5%, and 10% PPFR were found to be 340![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 237000 and 122000 g mol−1, respectively. The copolymer produced from 20% PPFR was oligomeric in nature with a molecular weight of 18000 g mol−1.
000, 237000 and 122000 g mol−1, respectively. The copolymer produced from 20% PPFR was oligomeric in nature with a molecular weight of 18000 g mol−1.
Introduction
Fluorinated polymers are niche macromolecules that play an integral role in modern life.1 They range from semi-crystalline to fully amorphous, and their uses span engineering thermoplastics and elastomers for the automotive and aeronautics industries, weather-proof coatings, biomedical materials, membranes for use in Li-batteries and fuel cells, and many more.2,3 Fluoropolymers possess exceptional physical and chemical properties, viz. high chemical-, thermal-, aging-, and weather resistance, as well as outstanding inertness to hydrocarbons, acids, and bases. Other highly desirable properties include low dielectric constant, low surface energy (water and oil repellency), low flammability, low refractive index, and low moisture absorption. Furthermore, the high strength of the C–F bond (485 kJ mol−1) grant fluoropolymers unparalleled resistance to oxidation.4–7Fluoropolymers also have their drawbacks. High crystallinity (up to 98% for PTFE)1,2,6 is often encountered in the homopolymers, resulting in poor solubility or total insolubility in common organic solvents and sparse solubility in fluorinated solvents. The poor solubility of fluorinated polymers and copolymers along with the general rigidity of their chains complicate the determination of molecular weight by the usual method of size exclusion chromatography (SEC).
The synthesis of fluorinated copolymers specially designed to overcome the problematic properties of the homopolymers has been the focus of extensive research.4,8,9 Recently Patil et al.10 utilised the persistent perfluoro-3-ethyl-2,4-dimethyl-3-pentyl radical (abbreviated as PPFR,11 structure shown in Fig. 1) to generate ˙CF3 radicals for polymerisation initiation. The resulting CF3 end groups were used in 19F NMR spectroscopy for the determination of average molecular weights of VDF copolymers. The technique provides Mn values closer to the true molecular weight of the copolymers than SEC. In addition CF3 labelling allows for the identification of the attack preferences of radical initiators on both co-monomers.
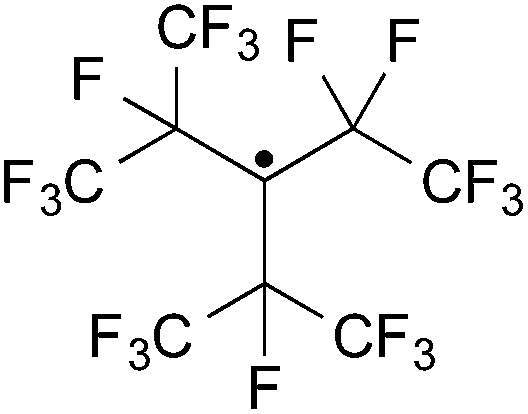 | ||
| Fig. 1 Structure of the perfluoro-3-ethyl-2,4-dimethyl-3-pentyl persistent radical (PPFR). | ||
In the research reported here, the use of PPFR as initiator was extended to the copolymers of CTFE and isobutyl vinyl ether. The molecular weights as well as the attack preferences of ˙CF3 onto the co-monomers, and the polymer thermal properties have been studied.
Experimental
Materials
Chlorotrifluoroethylene ≥99% (CTFE, CAS no. 79-38-9) was kindly provided by Honeywell (Buffalo, USA) and used as received. Isobutyl vinyl ether 99% (iBuVE, CAS no. 109-53-5), potassium carbonate 99.99% (K2CO3, CAS no. 584-08-7), dimethyl carbonate 99% (DMC, CAS no. 616-38-6), methanol ≥99.8% (CH3OH, ACS reagent, Ph. Eur., CAS no. 67-56-1) and acetone ≥99.5% (CH3COCH3, ACS reagent, Ph. Eur., CAS no. 67-64-1) were purchased from Sigma-Aldrich (Saint Quentin-Fallavier, France) and used as received. Distilled water was provided by an in-house purification system.The PPFR was kindly supplied by Prof. Taizo Ono of the Research Institute of Instrumentation Frontier in Nagoya. The PPFR was prepared by direct fluorination of a hexafluoropropene trimer precursor mixture at room temperature using undiluted fluorine gas. The PPFR solution was washed with 1 M aqueous Na2CO3 followed by distilled water, and then distilled under reduced pressure (25 mmHg). The distillate fraction boiling at 31–33 °C was used for this investigation. More information on the synthesis of the persistent radical may be found in Scherer et al.11
Polymerisation apparatus
The polymerisation reactions were conducted in a Parr Instruments (Moline, Illinois) stirred reactor (Hastalloy HC76). The reactor was equipped with an inlet valve and two outlet valves, a 3000 PSI rupture disc, a Span bourdon type pressure gauge and a thermowell. Temperature control was achieved by way of a heating jacket connected to a PID controller. A stainless steel sheathed (isolated) K-type thermocouple was used to monitor the temperature in the reactor.Radical polymerisation procedure
The reactor was subjected to 24 hours of acetone wash at 90 °C before each polymerisation run and pressure tested at 20 bar nitrogen before being subjected to high vacuum for 1 hour.The reaction mixture was prepared by dissolving 17.2 g of iBuVE and 0.0237 g of K2CO3 into 25 mL of DMC in a 50 mL round-bottom flask, with the PPFR initiator (1%, 5%, 10% and 20%, molar basis with respect to total monomer charge) being added after the potassium carbonate had dissolved. The reaction mixture was degassed under nitrogen for 20 minutes using the balloon and septum method. The reaction mixture was introduced to the reactor via a funnel tightly attached to the inlet valve with 25 mL of DMC used to wash out the degassing flask, ensuring that all the reagents were transferred to the vessel.
The reactor was immersed in liquid nitrogen until the DMC turned solid before the CTFE was transferred into the reactor. The mass of CTFE in the reactor was determined by weight difference (accurate to 0.5 g). The mass of CTFE was kept constant at 20 g in all the experiments.
The loaded reactor was left in a fume hood to warm to 25 °C over a 1 hour period before it was placed in the reactor stand. The reaction temperature was increased in a stepwise fashion to avoid overheating by first heating to 40 °C, then to 60 °C, then 80 °C and finally to 90 °C, stirring all the while. The reactor was left at 90 °C for 24 hours and afterwards cooled to 25 °C using an ice bath before it was degassed and opened.
In all cases, addition of PPFR to the solution of iBuVE in DMC resulted in an immediate yellowing of the solution. This is due to interaction between the π-orbitals of the vinyl ether and the PPFR radical.12 This interaction was considered to have no effect on the polymerisation reaction.
Product purification
The product solution from the reactor was evaporated in a rotary evaporator, leaving behind a viscous material. The impure polymer material was thrice dissolved in acetone and evaporated, before being dissolved in sufficient acetone to produce a saturated solution which was precipitated by drop-wise addition into a flask of vigorously stirred, cold (∼0 °C) methanol. The precipitate was filtered off, washed with methanol, precipitated a second time, filtered off, washed with methanol and dried under high vacuum at 80 °C. For the 20% PPFR experiment, distilled water rather than methanol was used as precipitant, as the product material dissolved in methanol.The product from the 1% PPFR experiment was isolated as a hard, whitish, opaque solid. The products from the 5% and 10% PPFR experiments were isolated as elastomeric, yellow to light orange solids and the product from the 20% PPFR experiment was isolated as a dark brown wax.
NMR characterisation
The nuclear magnetic resonance spectra were recorded on a Bruker AC 400 using deuterated chloroform. Coupling constants and chemical shifts are given in hertz (Hz) and parts per million (ppm), respectively. 1H, 19F, and proton-decoupled 19F NMR were performed.The experimental conditions for recording 1H, (or 19F) spectra were: flip angle 90° (or 30°); acquisition time of 4.5 s (or 0.7 s, pulse delay of 2 s), 32 scans (or 1024 scans); and a pulse width of 5 μs for 19F NMR.
NMR samples were prepared by dissolving 100 mg of polymer material in 1 mL of CDCl3. The 19F molecular weights were calculated using a similar procedure as previously reported.13
Thermogravimetric analysis (TGA)
Thermogravimetric analyses under nitrogen were performed using a Perkin Elmer TGA 4000 coupled to a Perkin Elmer Spectrum 100 FTIR spectrometer. Polymer samples (∼50 mg) were heated from 25 °C to 600 °C at 10 °C min−1 in air flowing at a rate of 50 mL min−1. The IR spectra were recorded from 550 cm−1 to 4000 cm−1 every 6 seconds at a resolution of 4 cm−1. Thermogravimetric analyses under air were carried out on a TGA 51 apparatus from TA Instruments. Polymer samples (∼15 mg) were heated from 25 °C to 500 °C at 10 °C min−1 in air flowing at a rate of 50 mL min−1.Differential scanning calorimetry (DSC)
Polymer samples (∼10 mg) were subjected to two heat-cool cycles under a nitrogen flow of 50 mL min−1. The polymer samples were heated from 25 °C to 150 °C at 10 °C min−1, held isothermally at 150 °C for 5 min, cooled from 150 °C to −150 °C at 10 °C min−1, held isothermally at −150 °C for 5 min, subjected to another heat-cool cycle before being heated from −150 °C to 25 °C at 10 °C min−1. Tg values were determined as the inflection point in the heat capacity jump.Size exclusion chromatography (SEC)
Size exclusion chromatography was conducted using a GPC 50 from Polymer Labs (now Agilent) with Cirrus software, as well as an Agilent 1260 Infinity system equipped with a Varian 390 LC triple detection system. The two systems used 2 PL Gel Mixed C columns (200 < Mw < 20 M g mol−1) with tetrahydrofuran (THF) as the eluent at a flow rate of 1.0 mL min−1 at 35 °C. The RI and UV detectors were calibrated using polystyrene standards. The viscometry detector used a universal calibration. Samples were prepared by dissolving 15 mg of polymer into 3 mL of THF followed by filtering through a 20 μm commercial PTFE filter. Analyses were achieved by injection of 20 μL filtered solution (5 mg mL−1).Results and discussion
The experimental conditions for the radical copolymerisation of chlorotrifluoroethylene (CTFE) with isobutyl vinyl ether (iBuVE) using PPFR as initiator, along with the characterisation results, are summarised in Table 1. PPFR has a half-life of 1 hour at 100 °C,11 releasing ˙CF3 and a branched perfluorinated 2-pentene (A) (Scheme 1).| Experiment | Monomer CTFE + iBuVE (mol) |
|
Yield (%) | Mna (g mol−1) | Mnb (g mol−1) | DP | PDI | Td10%,c (°C) | Tgd (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a Number average molecular weight as determined by SEC.b 19F NMR.c Decomposition temperature at 10% mass loss in air.d Glass transition temperature. | |||||||||
| 1 | 0.171 + 0.171 | 1 | 64 | 85000 |
340000 |
1570 | 2.13 | 330 | 20 |
| 2 | 0.171 + 0.171 | 5 | 46 | 70000 |
237000 |
1100 | 1.58 | 340 | 23 |
| 3 | 0.171 + 0.171 | 10 | 58 | 66000 |
122000 |
560 | 1.62 | 344 | 24 |
| 4 | 0.171 + 0.171 | 20 | 20 | 59000 |
18000 |
84 | 2.19 | 322 | 23 |

 | ||
| Scheme 1 β-scission elimination mechanism for the generation of ˙CF3 from PPFR. | ||
The CF3 radical initiates the radical copolymerisation of CTFE with iBuVE from 90° (Scheme 2), taking into account that its half-life is 3 hours at this temperature. A 50 mol% feed of CTFE/iBuVE was chosen since the maximum rate of polymerisation is found at this ratio.14
 | ||
| Scheme 2 Expected copolymerisation reaction of CTFE and iBuVE initiated by ˙CF3 to yield a poly(CTFE-alt-iBuVE) alternating copolymer. | ||
NMR characterisation
The progression of 19F NMR spectra going from 1% to 20% PPFR is presented in Fig. 2 (full NMR spectra are available as ESI†). The CF3–CH2 signal is observed at −66 ppm in the 1% PPFR spectrum with no other signals observed in the CF3 range. Similarly for the 5% and 10% PPFR spectra only the signal at −66 ppm is noted. For the 20% PPFR spectrum five signals are observed, viz. four major signals at −66, −77, −78, and −83 ppm. There is also one minor signal at −73.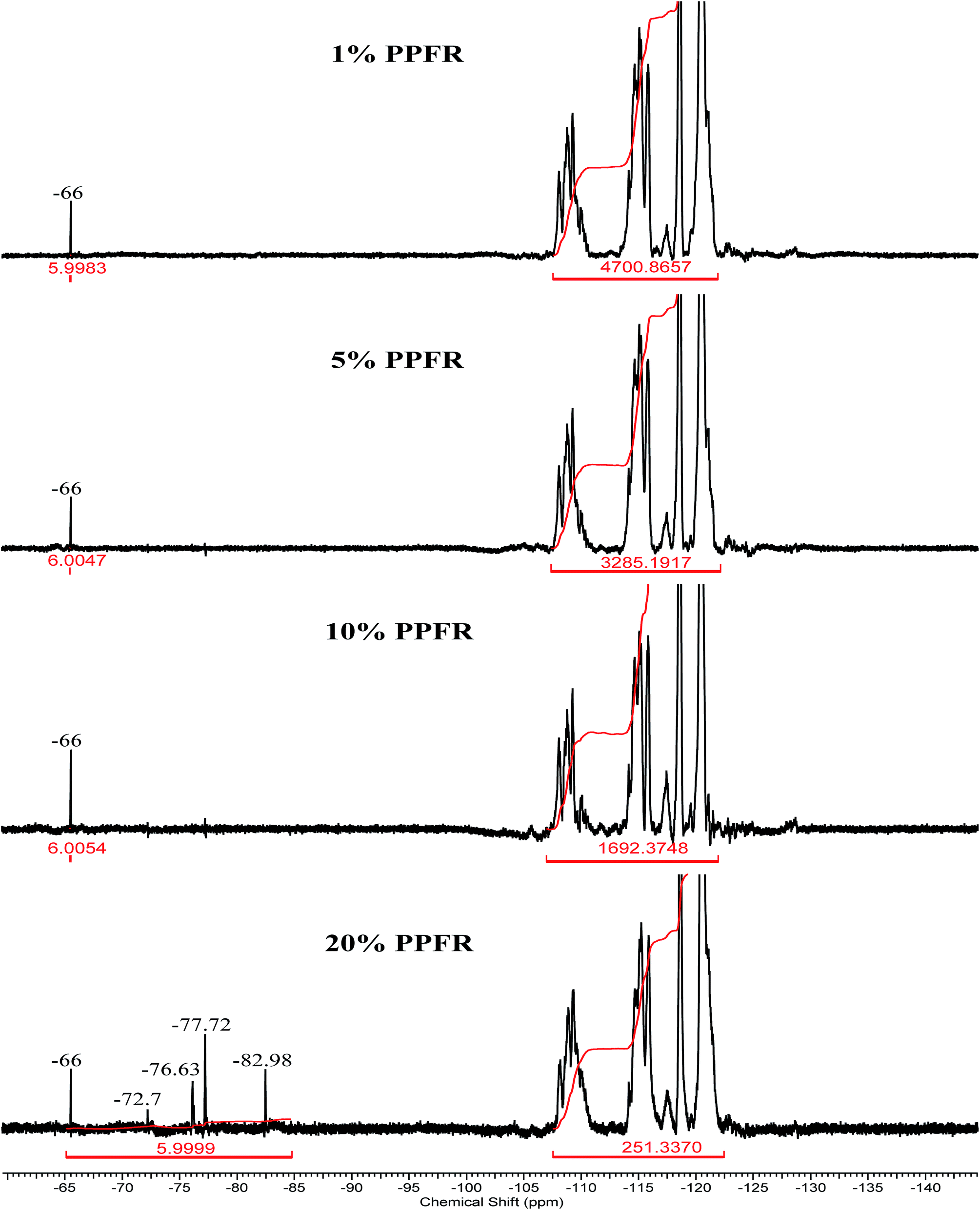 | ||
| Fig. 2 19F NMR spectra of poly(CTFE-alt-iBuVE) copolymers showing the progression of the CF3 19F NMR signal with increasing initiator concentration from 1 mol% (top spectrum) to 20 mol% (bottom spectrum). | ||
The 1H NMR spectra for 1% and 10% PPFR are presented along with that of pure iBuVE in Fig. 3. As expected from the literature,15,16 both the 19F and 1H NMR spectra displays signal broadening on the asymmetric carbons of CTFE and iBuVE units (in the range of −107 and −115 ppm for CTFE, and 4.5 ppm for iBuVE).
 | ||
| Fig. 3 1H NMR spectra of poly(CTFE-alt-iBuVE) copolymers at 1% and 10% PPFR concentration compared to that of isobutyl vinyl ether (top spectrum). | ||
The spectra agree with what is expected for poly(CTFE-alt-iBuVE) copolymer and demonstrates that the system produced via ˙CF3 radical initiation is alternating, as evidenced by the absence of any peaks in the −127 ppm range, indicative of CFCl groups in CTFE–CTFE dyads, in the 19F NMR spectrum.16–19
The five peaks present in the 19F NMR spectrum for 20% PPFR are unexpected. There are two plausible explanations: (1) the signals arise from ˙CF3 additions to carbon sites other than CH2; or (2) the signals arise from CTFE or iBuVE reaction with the PPFR elimination products of trans- and cis-perfluoro-3-ethyl-4-methyl-2-pentene.
The 1H NMR spectra (Fig. 3) of poly(CTFE-alt-iBuVE) exhibit the absence of signals centered at 6.53, 4.20, and 3.95 ppm characteristic of unreacted VE vinylic protons (upper spectrum). However, their polymerised unit can be found between 2.50 and 3.2 ppm, and between 4.3 and 4.7 ppm for the methylene and methyne protons, respectively.4,15,16,18,19
Both signals are broad arising from the presence of two types of asymmetric carbons leading to two diastereoisomers, which makes these protons anisochronous (i.e. nonequivalent). Methylene groups adjacent to the oxygen atoms, CH and both methyl groups in iBuVE are located at 3.45–3.65 ppm, 1.85 ppm, and 0.90 ppm, respectively.
There are several very small signals between 1.5 and 1 ppm. Several satellite signals near the signal assigned to the e carbons are observed in the neat iBuVE spectrum. The small signals in the polymer spectra are ascribed to cumulative intensities of the small signals seen in the iBuVE spectrum. These signals are due to 1H–13C coupling.
Addition preferences of ˙CF3 radicals to the CTFE/iBuVE charge transfer complex
The possible attack patterns of ˙CF3 on the CTFE/iBuVE acceptor–donor complex5,17,20 are presented in Scheme 3. Of these possible addition pathways, path 1a is considered the most likely as the highly electrophilic ˙CF3 radicals should preferentially attack the most electron donating, least hindered site in the monomer mixture.10,21–23 In the case of the vinyl ether, known to be a donating monomer,24 the preferred site will be the CH2 carbon, as it is not sterically hindered and is electron rich. | ||
| Scheme 3 Possible addition reactions of ˙CF3 radicals onto the CTFE/iBuVE.17,20 | ||
The literature10,13 indicates that, if the polymerisation occurs in a regioselective way via path 1a, there should be only one 19F NMR signal for CF3, centered at −63 ppm.
Works done on the telomerisation of CTFE with CF3CFClI25,26 showed that the signals for CF3 should be in the region of −77 ppm if it forms part of the CF3CFClCF2 motif and should be in the region of −82 ppm if it forms part of the CF3CF2CFCl endgroup. If the other pathways (such as attack onto the CTFE) are sufficiently viable, then there should be 19F NMR signals at around −77 to −78 ppm as well. The assignments of the signals in the 20% PPFR spectrum and their relative abundance are presented in Table 2.
| Chemical Shift (ppm) | Structural Assignmenta | Relative Abundance (%) |
|---|---|---|
a Assignments based on the NMR spectra for the telomerisation of CF2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CFCl with CF3CFClI and considerations from the literature.25,27 CFCl with CF3CFClI and considerations from the literature.25,27 |
||
| −66 | CF3–CH2–CH(O–iBu)–CF2–CFCl–R | 6 |
| −73 | CF3–CH(O–iBu)–CH2–CF2–CFCl–R | 5 |
| −77 | CF3–CFCl–CF2–CH(O–iBu)–CH2–R | 37 |
| −78 | CF3–CFCl–CF2–CH2–CH(O–iBu)–R | 47 |
| −83.0 | CF3–CF2–CFCl–R | 5 |
The relative abundances were calculated by initially setting the integration of the −66 ppm signal to 6 and then dividing the integral of each CF3 signal by the sum of the integrations of all the CF3 signals.
Assuming that the signals in the 19F NMR spectrum for 20% PPFR are only due to different ˙CF3 additions, then the NMR results show that at low initiator concentrations, pathway 1a is favoured, but at higher initiator concentrations pathway 2a becomes dominant. The low prevalence of attack via 2b is due to electronic effects, the CF2 carbon being electron poor. The low occurrence of attack via 1b is due to steric and electronic effects.
The effect of initiator concentration is remarkable in that there are no electronic or steric considerations that shift the regioselectivity away from CH2 towards CFCl attack. The previous work with VDF and PPFR was done in halogenated solvents, so the current reaction behaviour is possibly governed by kinetic effects arising from solubility considerations.
PPFR is a fully fluorinated species which dissolves in dimethylcarbonate (DMC) at low concentrations. However, at higher concentrations, a two phase system exists at standard conditions. The phase behaviour of large, sterically hindered, fully fluorinated molecules in contact with DMC at elevated temperatures and pressures is not known, but it is suspected that there exists, under the reaction conditions used here, a two phase system, one phase being DMC rich and the other being fluorous.
The solubility of CTFE in the fluorous phase should be an order of magnitude higher than its solubility in DMC. The “regioselectivity” at higher initiator concentrations is then ascribed simply to the much higher abundance of CTFE over iBuVE near to the initiator molecules.
Reaction of iBuVE with PPFR elimination products
At low initiator concentrations, any effect of the elimination products on the reaction is overshadowed by the much greater abundance of other monomers. However, at concentrations of 20% PPFR, the molar quantities of unsaturated fluorinated elimination product cannot be ignored when interpreting the results.The decomposition reaction of PPFR is shown in Scheme 1. The by-product from ˙CF3 elimination is a sterically hindered, unsaturated fluorocarbon. Normally, such hindered fluorocarbons do not homopolymerise under radical conditions, as is the case with hexafluoropropylene (HFP) or perfluoroalkylvinyl ethers (PAVEs). However, it has been shown that even HFP and PAVEs readily produces alternating copolymers with vinyl ethers.28,29 Accordingly, there exists a distinct possibility that the iBuVE may react with the unsaturated elimination A product to produce a copolymer. A possible reaction is shown in Scheme 4.
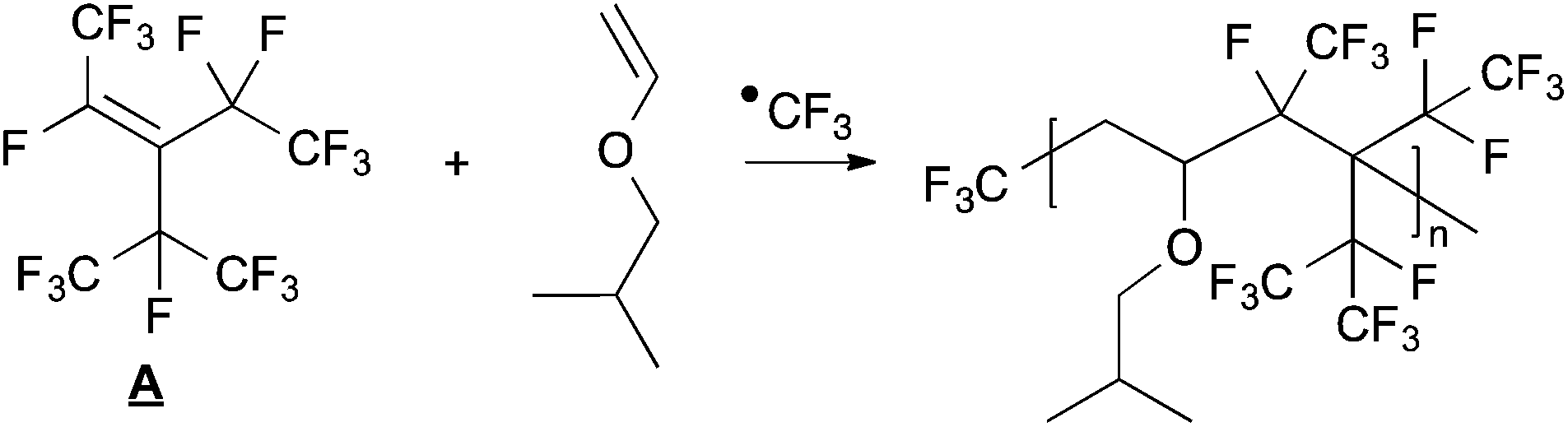 | ||
| Scheme 4 Possible polymerisation reaction between iBuVE and perfluoroolefin A. | ||
However, the terminal radical that would form on product A in such a reaction should be unreactive as its environment is sterically very similar to the radical centre of PPFR. A much more plausible scenario is the addition of iBuVE onto A to form a stable, radically capped monoadduct instead of a polymer.
If such a product did exist there should be 19F NMR signals at around −175 ppm. Since no such signals are detected, this reaction did not occur and the unexpected 19F NMR signals for CF3 observed in the 19F NMR spectrum for 20% PPFR cannot be due to incorporation of the PPFR elimination product into the polymer.
Effect of initiator concentration on molecular weight
The normalised size exclusion chromatograms (SEC or GPC) are displayed in overlaid form in Fig. 4. The expected decrease in molecular weight with increasing initiator ratio is noted. However, the concentration of initiator is observed to have limited effect on molecular weight beyond 5%, displaying a significant drop in molecular weight from 1% to 5% (with Mn ranging from 85000 to 70000 g mol−1), but showing a nearly linear correlation for decreasing molecular weight with increase of the square root of the ratio of initiator concentration to the monomer concentration from 5% to 20% (with Mn ranging from 66000 to 59000 g mol−1). This trend is presented graphically in Fig. 5.
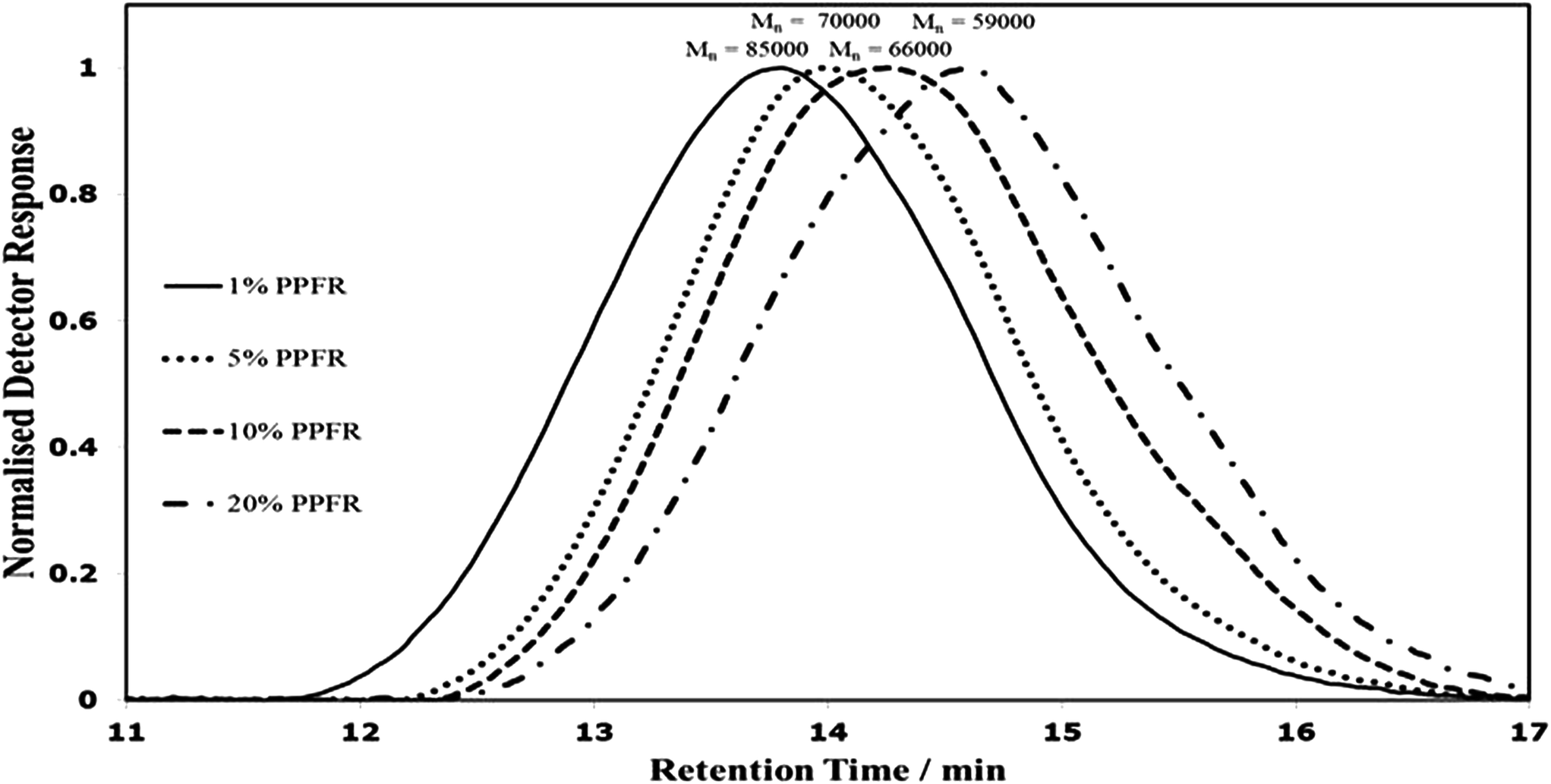 | ||
| Fig. 4 Size exclusion chromatograms showing the number average molecular weights of poly(CTFE-alt-iBuVE) copolymers prepared from various amounts of PPFR radical molar percentages. | ||
 | ||
| Fig. 5 Correlation of Mn decrease as PPFR ratio increases as determined by both SEC (△) and 19F NMR spectroscopy (■). | ||
The molecular weights determined by 19F NMR also show the expected decrease in molecular weight with increase in initiator concentration, with the trend being nearly linear in the region of 1% to 10% initiator (with Mn ranging from 340000 to 122000 g mol−1).
As expected, since the GPC standards are polystyrene, there is a very large difference between the Mn values derived from SEC (or GPC) and NMR.
Thermal properties of CF3 terminated poly(CTFE-alt-iBuVE) copolymers
 | ||
| Fig. 6 TGA thermograms for the poly(CTFE-alt-iBVE) copolymers under N2 at 10 °C min−1. | ||
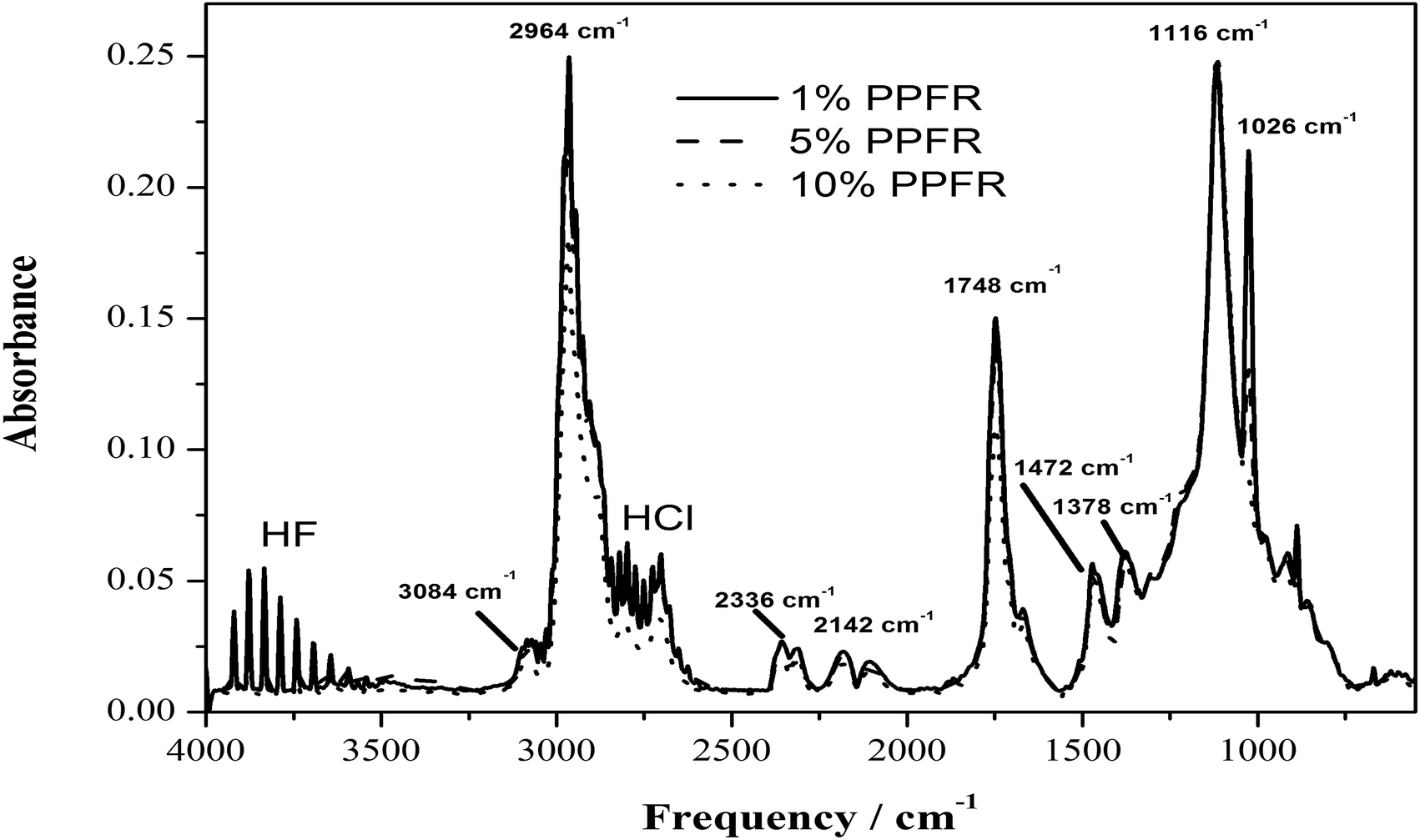 | ||
| Fig. 7 FTIR spectrum of the evolved gases from the thermal decomposition of the poly(CTFE-alt-iBuVE) alternating copolymers, taken at the point of maximum absorbance, showing, among other species, the evolution of HF, HCl, CO and CO2. | ||
The thermal degradation behaviour and degradation mechanism of fluoropolymers are dependent on the chemical nature of the polymer, the chain length and the morphology of the chains.30 For fully fluorinated polymers, the usual degradation routes involve either unzipping from the chain ends or breakdown due to random chain scission while for partially fluorinated polymers, dehydrofluorination is usually the main mechanism of degradation.31,32 Polymers synthesised via non-fluorinated initiators are highly susceptible to unzipping from the chain ends or to oxidative attack initiated at the non-fluorinated chain ends.
The use of CF3 as terminal group enables the gauging of the degradation behaviour of poly(CTFE-alt-iBuVE) free from breakdown initiated at the chain ends.
The thermograms show that the polymer produced with 1 mol% PPFR is the most stable, with a degradation onset temperature of 387 °C, as is expected considering that its molecular weight is the largest of all the polymers. Interestingly, the 5 mol% and 10 mol% polymers show nearly the same degradation onset temperature (367 °C). The 20 mol% polymer is not shown as the polymer underwent significant mass loss, via evaporation of the low molecular weight chains, long before the other polymers started degrading, with the onset of loss temperature for the higher molecular weight fraction of the 20 mol% PPFR polymer being in the region of 325 °C.
The 10% mass loss temperature under air is reported in Table 1, and demonstrates that the thermal behaviour of the 5 mol% and 10 mol% polymers are nearly identical under both air and nitrogen. The differences in onset temperatures between heating in air and in inert atmosphere demonstrates that the alternating poly(CTFE-alt-iBuVE) copolymer undergoes combustion long before the polymer chains start degrading, implying that in the event of combustion during service, the subsurface polymer material will remain in serviceable condition when the surface combustion is extinguished. Also, the differences in degradation onset temperatures between the 1 mol% PPFR, 5 mol% and 10 mol% PPFR polymers demonstrates that the degradation behaviour is dependent on molecular weight.
Furthermore, these results indicate that the intrinsic oxidative thermal stability of the poly(CTFE-alt-iBVE) backbone is quite good for a partially fluorinated polymer, being comparable to the stability achieved by fully fluorinated polymers. For comparison, the onset of degradation temperature for PTFE is around 550 °C (ref. 33) under the same conditions.
The infrared spectra of the evolved gases display the expected release of HF and HCl. The infrared absorbance bands for CO and CO2 are also observed, at 2142 and 2336 cm−1, respectively. The peak at 1748, 3084, 2964 and 1116 cm−1 is indicative of a CO group, a CC–H stretch, a CH2 stretch and of C–H bends, respectively. Also, no peaks for an O–H bond, usually seen at around 3600 cm−1 are observed.
While the gas phase is obviously a mixture of species, the FTIR spectra indicate that the major component of the organic pyrolysis products is something akin to isobutenal. Given that CO and CO2 are also produced, other minor components like isobutylene and ethers of isobutene must also be present in the gas phase. The literature31,32 indicates that monofluorides and monochlorides of these compounds should also be present in some small quantity.
The work of Zulfiqar et al.31,32,34 focused on the random copolymers of CTFE with methyl methacrylate, VDF and styrene. These polymers are all random copolymers. It was noticed there that the degradation of CTFE–MMA copolymers occurs in a two-step process, with HCl being eliminated first, followed by depolymerisation of the polymer chain to MMA and chlorofluorocarbons. No mention is made of the elimination of HF.
The thermal decomposition of polyvinyl alcohol and polyvinyl acetate (model polymers approximating vinyl ethers) also occur in a two-step process. Polyvinyl alcohol first releases water, forming double bonds in the polymer chain and afterward undergoing chain scission to produce unsaturated aldehydes and ketones. Poly vinyl acetate first releases acetic acid to form an unsaturated, ketonic polymer backbone, followed by elimination of ketones from the unsaturated chain.35,36
In the case of poly(CTFE-alt-iBuVE), the breakdown is seen to be one step with HCl, HF and the pendant ether group eliminated from the polymer chain concurrently, with minimal internal rearrangements and reactions of the pendant ether groups with the radicals in the polymer backbone.
:1 ratio of CTFE:iBuVE.Conclusions
A series of poly(CTFE-alt-iBuVE) alternating copolymers has been synthesised in good yield via radical polymerisation initiated by ˙CF3 released from the β-scission of perfluoro-3-ethyl-2,4-dimethyl-3-pentyl persistant radical at 90 °C. The addition behaviour of ˙CF3 radicals onto the CTFE/iBuVE is highly dependent on the initiator-to-monomer ratio. It was demonstrated that ˙CF3 radicals preferentially attack the methylene site in the vinyl ether monomer to initiate chain cross propagation when using low initiator concentrations. At initiator concentrations of 20% it was shown that there is significant deviation from the expected behaviour, ascribed to either the effects of initiator. The usefulness of CF3 end groups as labels for molecular weight determination in poly(CTFE-alt-iBVE) copolymers by 19F NMR was demonstrated and compared to results obtained by SEC. The polymerisation initiated by PPFR persistent radical exhibiting the expected tendency for the molecular weight of the copolymers to increase with decreasing initiator concentration.Acknowledgements
The authors thank the National Research Foundation of South Africa, CNRS, France and the south African Department of Science and Technology's Fluorochemical Expansion Initiative for the opportunity to collaborate and for funding this research, and Honeywell company for supplying the CTFE.Notes and references
- J. G. Drobny, Technology of Fluoropolymers, CRC Press, Boca Raton, 2014 Search PubMed.
- S. Ebnesajjad, Fluoroplastics, Volume 1: Non-Melt Processible Fluoroplastics, Elsevier Science, Amsterdam, 2000 Search PubMed.
- S. Ebnesajjad, Fluoroplastics, Volume 2: Melt Processible Fluoroplastics: The Definitive User's Guide, Elsevier Science, Amsterdam, 2002 Search PubMed.
- F. Boschet and B. Ameduri, Chem. Rev., 2014, 114, 927–980 CrossRef CAS PubMed.
- B. Ameduri and B. Boutevin, Well-Architectured Fluoropolymers: Synthesis, Properties and Applications: Synthesis, Properties and Applications, Elsevier Science, 2004 Search PubMed.
- G. Hougham, P. E. Cassidy, K. Johns and T. Davidson, Fluoropolymers 1, Springer, US, New York, 1999 Search PubMed.
- D. W. Smith, S. T. Iacono and S. S. Iyer, Handbook of Fluoropolymer Science and Technology, Wiley, New York, 2014 Search PubMed.
- B. Ameduri, Chem. Rev., 2009, 109, 6632–6686 CrossRef CAS PubMed.
- Y. Patil, T. Ono and B. Ameduri, ACS Macro Lett., 2012, 1, 315–320 CrossRef.
- Y. Patil, A. Alaaeddine, T. Ono and B. Ameduri, Macromolecules, 2013, 46, 3092–3106 CrossRef CAS.
- K. V. Scherer, T. Ono, K. Yamanouchi, R. Fernandez and P. Henderson, J. Am. Chem. Soc., 1985, 107, 718–719 CrossRef CAS.
- T. Ono and K. Ohta, J. Fluorine Chem., 2014, 167, 198–202 CrossRef CAS PubMed.
- F. Boschet, T. Ono and B. Ameduri, Macromol. Rapid Commun., 2012, 33, 302–308 CrossRef CAS PubMed.
- Y. Tabata and T. A. Du Plessis, J. Polym. Sci., Part A-1: Polym. Chem., 1971, 9, 3425–3435 CrossRef CAS PubMed.
- G. Couture, B. Campagne, A. Alaaeddine and B. Ameduri, Polym. Chem., 2013, 4, 1960–1968 RSC.
- A. Alaaeddine, G. Couture and B. Ameduri, Polym. Chem., 2013, 4, 4335–4347 RSC.
- G. V. D. Tiers and F. A. Bovey, J. Polym. Sci., Part A: Gen. Pap., 1963, 1, 833–841 CrossRef CAS PubMed.
- M. Gaboyard, Y. Hervaud and B. Boutevin, Polym. Int., 2002, 51, 577–584 CrossRef CAS PubMed.
- D. Carnevale, P. Wormald, B. Ameduri, R. Tayouo and S. E. Ashbrook, Macromolecules, 2009, 42, 5652–5659 CrossRef CAS.
- Z. M. O. Rzaev, Prog. Polym. Sci., 2000, 25, 163–217 CrossRef CAS.
- Y. Takeyama, Y. Ichinose, K. Oshima and K. Utimoto, Tetrahedron Lett., 1989, 30, 3159–3162 CrossRef CAS.
- J. N. Cape, A. C. Greig, J. M. Tedder and J. C. Walton, J. Chem. Soc., Faraday Trans. 1, 1975, 71, 592–601 RSC.
- K. Miura, Y. Takeyama, K. Oshima and K. Utimoto, Bull. Chem. Soc. Jpn., 1991, 64, 1542–1553 CrossRef CAS.
- D. J. T. Hill, J. J. O'Donnell and P. W. O'Sullivan, Prog. Polym. Sci., 1982, 8, 215–275 CrossRef CAS.
- M. P. Amiry, R. D. Chambers, M. P. Greenhall, B. Ameduri, B. Boutevin, G. Caporiccio, G. A. Gornowicz and A. P. Wright, The peroxide initiated telomerization of Chlorotrifluoroethylene with perfluorochloroalkyl iodides, Polym. Prepr., 1993, 34, 411–412 CAS.
- R. N. Haszeldine, J. Chem. Soc., 1955, 4291–4302 RSC.
- W. R. Dolbier, Guide to Fluorine NMR for Organic Chemists, Wiley, New York, 2009 Search PubMed.
- D. Valade, F. Boschet and B. Améduri, Macromolecules, 2009, 42, 7689–7700 CrossRef CAS.
- A. Alaaeddine, F. Boschet, B. Ameduri and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3303–3312 CrossRef CAS PubMed.
- T. R. Crompton, Thermo-oxidative Degradation of Polymers, iSmithers, Akron, Ohio, 2010 Search PubMed.
- S. Zulfiqar, M. Rizvi, A. Munir, A. Ghaffar and I. C. McNeill, Polym. Degrad. Stab., 1996, 52, 341–348 CrossRef CAS.
- S. Zulfiqar, M. Zulfiqar, M. Rizvi, A. Munir and I. C. McNeill, Polym. Degrad. Stab., 1994, 43, 423–430 CrossRef CAS.
- G. J. Puts and P. L. Crouse, J. Fluorine Chem., 2014, 168, 260–267 CrossRef CAS PubMed.
- S. Zulfiqar, M. Rizvi and A. Munir, Polym. Degrad. Stab., 1994, 44, 21–25 CrossRef CAS.
- B. J. Holland and J. N. Hay, Polymer, 2002, 43, 2207–2211 CrossRef CAS.
- B. J. Holland and J. N. Hay, Polymer, 2001, 42, 6775–6783 CrossRef CAS.
- J. W. Nicholson, The Chemistry of Polymers, Royal Society of Chemistry, London, 2012 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c5ra05066a |
| This journal is © The Royal Society of Chemistry 2015 |