
Hydrothermal synthesis of nitrogen doped graphene nanosheets from carbon nanosheets with enhanced electrocatalytic properties†
Abstract
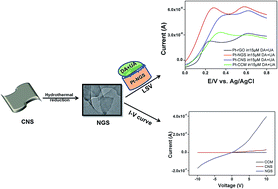
Synthesis of nitrogen doped graphene nanosheets (NGS) with controlled structure is a burgeoning issue in the field of materials chemistry. Herein, we for the first time report a novel, green, low-temperature, hydrothermal synthesis of two dimensional (2D), porous, NGS from carbon nanosheets (CNS) as precursor materials. Hydrothermal reduction of CNS not only imparts crystallinity to the material but also results in a substantial removal of oxygen functionalities, forming NGS. The as-synthesized NGS displayed excellent electrochemical sensing properties with high selectivity and sensitivity for the detection of dopamine (DA) in human urine samples. NGS can easily distinguish the electrochemical oxidation peak for DA from that of uric acid (UA), which is a common interferent for DA detection. The electrochemical oxidation peak current of DA linearly varies within the concentration range of 0.1 to 100 μM, having remarkable sensitivity (2.661 μA μM−1) and a lower detection limit upto 100 nM, with a correlation coefficient (R2) of 0.9880. The separation of electrooxidation peak potential for DA–UA is extraordinarily high (0.37 ± 0.05 V). In contrast to carbonized carbon material (CCM), reduced graphene oxide (rGO) and other carbon materials, the current response for NGS is three folds higher at the same DA concentration with higher electrocatalytic response. Furthermore, superior electrochemical sensitivity and selectivity along with enhanced electrocatalytic properties of NGS for DA detection is metal/metal oxide free, which are usually employed for electrode modification to achieve higher current response. Henceforth, we believe that NGS synthesis offers great promises for creating a revolutionary new class of nanostructured electrodes for biosensing applications.
 Please wait while we load your content...
Please wait while we load your content...

