
Liquid–solid transition in mesophase separated olefin multiblock copolymers during crystallization
Peng He,
Bin Chen,
Wei Yu* and
Chixing Zhou
Advanced Rheology Institute, Department of Polymer Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, P. R. China. E-mail: wyu@sjtu.edu.cn
First published on 29th April 2015
Abstract
The effect of block structure on the liquid–solid transition (LST) of ethylene–octene multiblock copolymers (OBCs) during isothermal crystallization has been investigated by rheology, differential scanning calorimetry (DSC), and polarized optical microscopy (POM). Due to the mesophase separation in OBC melts, the formation of a critical network at the LST in the OBCs with low crystallinity (7–14 wt%) was found to be different from that in homogeneous systems. The viscoelastic properties at the LST in the heterogeneous OBCs suggested a slower relaxation behavior of the critical network, and the liquid–solid transition in strongly segregated OBCs was observed to occur in the intermediate or even late stage of crystallization, demonstrated by the much higher crystallinity and large spherulites at the LST. The delayed liquid–solid transition has been discussed and can be attributed to the initial confinement of the hard-block domains in the nucleation and growth of the crystals.
1. Introduction
Polymer crystallization has been the subject of intense research over the past years since it is of great importance to material properties and practical processing.1–3 It was shown that the liquid–solid transition during crystallization can be viewed as a physical gelation process, and the crystallites act as the physical crosslinks to connect the molecular chains.3–6 The critical transition point (or the critical gel point), which marks the transition from liquid-like state to solid-like state, can be defined as the appearance of sample spanning connectivity. Compared to other techniques, rheological methods have been demonstrated as sensitive tools to probe the subtle structure changes at the liquid–solid transitions (LST).6In the past decades, the gelation concept has been applied successfully to investigate the chemical-crosslinking7–9 and the crystallization process3–5,10–12 in various polymer systems. The work on the crystallizing polymers have generally focused on the time scales of the gelation process,4,10 the rheological properties of the critical gel,11 and the crystallinity at the gel point.3,5,12 An important question is the correlation between the viscoelastic properties and the connecting structure of the network at LST for a crystalline polymer. Several interpretations3,5,6,13 had been proposed based on the rheological properties at the critical point. Horst and Winter5 investigated the critical gel properties of ethylene–butene random copolymer and discussed several possibilities for the critical network, i.e., the instant impingement between the crystallites, a network of amorphous chains connected by small crystallites, and a network of bridging chains embedded in several large crystalline regions, similar to the network formed by chemical cross-linking. Acierno et al.13 suggested that the formation of the critical network in crystallizing poly(1-butene) resembles the generation of multi-arm polymer from a matrix of linear chains. Coppola et al.3 further investigated the same polymer system by Acierno,13 and found that the amorphous segments of copolymer plays an important role in the connectivity among crystallites because of a long relaxation process similar to that in hybrid colloid-polymer systems. The consensus of these work is that the connectivity of the superstructure at the critical point can be achieved by the coupling effect of amorphous chains and the very low amount of crystallites originated from the homogeneous polymer melts. However, if the crystallization occurred from the microphase separated melts of semicrystalline block copolymers, the competition of the microphase separation and crystallization would complicate the formation of critical network, which possibly possesses viscoelastic properties distinct from that in homogenous systems.
Chain shuttling polymerization14 has recently enabled the efficient production of olefin multiblock copolymers (OBCs) with a linear structure of alternating hard segments (crystallizable ethylene–octene blocks with low octene content) and soft segments (amorphous ethylene–octene blocks with high octene content). This novel block structure imparts excellent elastomeric properties at higher temperatures than the random ethylene–octene copolymer.15 The high elasticity of OBCs can arise from the crystallization of the hard segments,16–19 which form physical crosslinks between the soft segments. For semicrystalline OBCs, both confined crystallization and breakout crystallization had been found, depending on the competition between the mesophase separation in the melt and crystallization.20–27 For weakly melt segregated OBCs, crystallization breaks out from the mesophase separated structures.19,21 For strongly melt segregated OBC, mesophase separation can be maintained and stacks of lamellae are confined within the isolated domains.21,23,25 However, larger crystals across domains had also been observed in strongly segregated OBCs with sphere25 or lamella22,24 mesophase structures. It was explained from the inter-block mixing due to the polydispersity of block length.22 Short hard blocks dissolved in the soft blocks allow hard block crystallization to proceed across the lamellar domain interfaces, leading to a new mode of crystallization, termed as “pass through”.22 Considering the different pathways of crystallization in OBCs, the raised question is that whether the connectivity and the rheological properties of the network during liquid–solid transition are influenced by the melt structure of OBCs. The understanding of them are very important to their practical application of thermoplastic elastomers.
In the preceding work,26 we have explored the melt-phase behaviors of a series of multi-block OBCs with varying block structures. Based on the understanding of the melt structures, the present work will study the liquid–solid transitions of OBCs during crystallization. Compared to those studies on the confined crystallization of OBCs at the nanoscale,21,23–25 the main goal of this work is to clarify the influence of melt structure on the large-scale network formation during crystallization.
2. Experimental
2.1 Materials
The OBCs synthesized by the chain-shuttling technology14 were supplied as pellets by The Dow Chemical Company. In this work, five OBCs with different block structures were chosen for comparison. The information about the molecular structure, and the transition temperatures for OBCs are provided in Table 1. The octene content in the hard and soft block of these OBCs are all controlled to be around 1.0 mol% and 18.0 mol%, respectively. The differences between them are mainly the hard block content (fhard) and molecular weight (Mw). The OBCs are designated as Lx or Mx, where L(M) denotes the low(medium) fraction of hard segments, and x is number that reflecting the relative level of molecular weight. More detailed characterization and discussion on the block structures of these OBCs can be found in a previous work.26| Sample code | Mw (kg mol−1) | PDI | fhard (wt%) | Total C8 (mol%) | Tm,peaka (°C) | Tm,comb (°C) | Tc,onsetc (°C) | Tc,peakd (°C) | TMSTe (°C) | Xc,ΔHf (wt%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a The melting peak temperatures.b The melting completion temperatures.c The crystallization onset temperatures.d The crystallization peak temperatures.e The Mesophase separation transition temperatures from rheology.f Crystallinity from heat of melting. | ||||||||||
| L01 | 79.8 | 1.97 | 11.0 | 13.7 | 122.8 | 126.3 | 102.0 | 94.6 | 163.5 | 7.4 |
| L02 | 126.5 | 2.06 | 11.0 | 14.0 | 120.7 | 125.9 | 97.1 | 89.9 | >200 | 7.0 |
| M01 | 61.7 | 1.83 | 25.0 | 11.7 | 122.9 | 126.8 | 106.2 | 101.0 | 153.8 | 14.0 |
| M02 | 82.6 | 2.15 | 25.0 | 12.0 | 122.8 | 128.6 | 103.1 | 95.9 | 160.4 | 14.8 |
| M03 | 156.6 | 1.97 | 25.0 | 11.5 | 119.7 | 124.6 | 100.0 | 96.3 | >200 | 14.3 |
2.2 Measurements
The rheological data were all acquired from the small amplitude oscillatory shear (SAOS) measurements with parallel plate fixtures (diameter 25 mm). A stress-controlled rheometer (AR-G2, TA instrument) was used to measure rheological properties of OBCs in the molten state. The dynamic frequency sweeps (100–0.01 rad s−1) were performed under different temperatures at a strain amplitude of 5%. A strain-controlled rheometer (ARES-G2, TA instrument) was used to monitor the nonisothermal and isothermal crystallization processes for OBCs under quiescent conditions. The nonisothermal crystallization processes were conducted by cooling from the melt at a rate of 5 °C min−1. In isothermal crystallization experiment, the sample was cooled rapidly from melt to the final crystallization temperature, Tc, at a rate of 20 °C min−1. Then, different rheological techniques were applied to track the isothermal crystallization processes, depending on Tc, as shown in Fig. 1. | ||
| Fig. 1 Schematic indication of quiescent crystallization processes of olefin block copolymers. (a) Cyclic frequency sweep at higher Tc, from 100 to 0.01 rad s−1, (b) time sweep at lower Tc, 1 rad s−1. | ||
At higher Tc, close to the melting point, the frequency sweeps from 100 to 0.01 rad s−1 were performed repeatedly to determine the critical LST point for OBCs (Fig. 1a). According to Winter's liquid–solid transition (LST) theory,6 the storage modulus (G′) and the loss modulus (G′′) from SAOS at the critical point follow the power law behavior:
 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ is frequency independent within the low frequency range at the critical point:
δ is frequency independent within the low frequency range at the critical point:
 | (2) |
It had been proved to be a reliable tool to rheologically determine the critical point during liquid–solid transition in many crystallizing polymers.3–5,10,13,28–34 However, the limitation of this method is that as the requirement of sufficiently low-frequency measurements, the experiments should be conducted at low undercoolings to follow the crystallization processes with slow crystallization rate, which make sure that no significant changes occur during single sweep. At lower Tc, as the crystallization rate is fast, the evolution of viscoelastic properties were measured until the late stage of crystallization through the oscillatory time sweep mode with an angular frequency of 1 rad s−1 and a strain amplitude of 2%, as shown in Fig. 1b.
Differential scanning calorimetry (DSC) measurements were conducted on Q2000 (TA Instruments) to investigate the isothermal crystallization of OBCs. The thermal treatments and the isothermal crystallization temperatures were the same as that in rheological measurements. The specimens were 4–6 mg cut from the OBC pellets and were sealed with aluminum pans. The instrument was calibrated with indium and tin. The isothermal crystallization were all carried out in nitrogen.
Polarized optical microscopy (POM) observations were carried out to investigate the crystalline morphology of OBCs. The experiments were performed on a Leica microscope (DM 2500P) and the experimental temperature was controlled by a Linkam hotstage. The thermal treatment and the isothermal crystallization temperatures were also the same as that in rheological measurements. The specimens for POM observation were films of about 50 μm prepared by pressing the pellets at 190 °C between two glass slides, and slowly cooling down to ambient temperature.
3. Results and discussion
3.1 Mesophase separation in molten state
In the preceding work,26 the melt phase behaviors of five OBCs had been well investigated. The determined mesophase separation temperatures are shown in Table 1. In this work, our attention is focused on the crystallization of OBCs. Considering the importance of the influence of mesophase on the crystallization behaviors, the vGP plots35 are given to simply depict the different melt structures of the five OBCs. The vGP plot (plotting phase angle, δ, versus the absolute value of the complex modulus, |G*|) is independent of temperature and can provide a way to check the validity of time-temperature-superposition (TTS) principle. It had been found to be an effective method to detect the liquid–liquid phase separation of polymer blends.36,37The vGP plots of the five OBCs at various temperatures are shown in Fig. 2. For M01, the vGP plots at different temperatures can superpose well onto a single curve, and show the typical properties of homogeneous polymer melt with phase angle approaching 90° at low |G*| values, which is equivalent to the terminal flow range in the usual dynamic modulus-frequency master curve and indicate viscous behavior.38 With the increase in molecular weight (M01 to M02), the vGP plots for M02 at high temperatures can superimpose perfectly, but slight deviations appear at low |G*| values at low temperature (135 °C), indicating the failure of TTS and a certain degree of mesophase separation in M02. Similar trend is seen in L01, whose molecular weight is close to M02. As for M03 and L02, which have the highest molecular weight in these OBCs, the vGP plots fail to superimpose within the experimental temperature range, indicating that the stronger mesophase separations occur in M03 and L02 and the homogeneity cannot be reached at temperature as high as 215 °C. These features from vGP plots for OBCs are in good agreement with the conclusion of the previous work.26 In general, the mesophase separation in OBCs can be controlled by increasing the Mw from L01 to L02 (similarly from M01 to M03). According to these results, we simply divide the five OBCs into several category: nearly homogeneous for M01, weakly mesophase separated for M02 and L01, stronger mesophase separated for L02, and the strongest mesophase separated for M03.
 | ||
| Fig. 2 The vGP plots, phase angle, δ, vs. the absolute value of the complex modulus, |G*| of OBCs from linear viscoelastic measurements at various temperatures. (a) M01, M02 and M03, (b) L01 and L02. | ||
3.2 Nonisothermal crystallization behaviors
The melting and crystallization temperatures of OBCs from nonisothermal DSC measurements are summarized in Table 1. It is noted that the melting temperature, Tm,peak, of L01, M01 and M02 are nearly at the same value of 122.8 °C, suggesting the melting behaviors of OBCs are less affected within such low Mw range. Obvious decreases can be found in high Mw OBCs (L02 and M03). In the random ethylene copolymers,39 Tm,peak had been found to increase with Mw increase. As the Mw was further increased above a critical value, the influence of Mw on Tm,peak is much less significant. This is explained that the Tm, peak of the random ethylene copolymers is determined by the longest ethylene sequence length. For the investigated OBCs, even though the Mw ranges from 61.7 kg mol−1 to 82.6 kg mol−1, the similar Tm,peak of the OBCs suggests that the crystallizable hard blocks are long enough to form well-organized crystals with high melting temperature, i.e., the longest crystallizable ethylene sequence lengths are similar in hard blocks within the Mw range. However, when the Mw are further increased (e.g. M02 to M03), the obvious decrease in both melting and crystallization temperatures could be related to the influence of the mesophase separation caused by increasing Mw, which needs to be carefully investigated.The evolutions of viscoelastic properties for OBCs during cooling are presented in Fig. 3. The liquid–solid transition during crystallization results in a steep increase in G′. In Fig. 3a, the overall crystallization transition temperature range for the five OBCs from rheology are consistent with the DSC results, and the minor differences are caused by the different cooling rates in rheology and DSC. With the similar hard block content (close crystallinity), the G′ curves for M01, M02 and M03 tend to superimpose after crystallization at lower temperature. Similar phenomenon can also be observed in OBC L-series with the similar hard block content of 11 wt%. The OBC M-series, with the higher block content, show the steeper moduli transitions and the higher modulus after crystallization than the OBC L-series. The above results suggest that the steepness of the modulus growth and the final modulus after crystallization are more dependent on block content (crystallinity), and is not much related to the Mw. For OBCs with the similar block content, the low Mw sample experiences larger change of modulus during the liquid–solid transitions, which can be ascribed to the low elasticity in melts. However, more information about the underlying structure changes cannot be obtained from the non-equilibrium states during non-isothermal conditions. The subsequent sections will focus on the effect of block structure on the isothermal crystallization of OBCs.
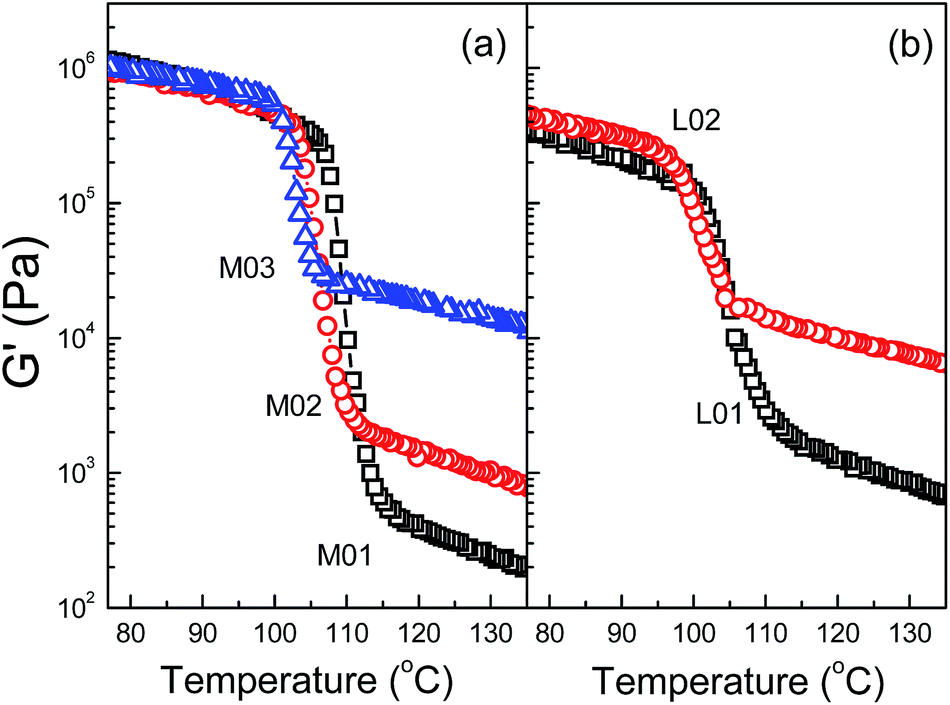 | ||
| Fig. 3 Evolution of dynamic modulus of OBCs during nonisothermal crystallization (a) M01, M02 and M03, (b) L01 and L02. (Cooling rate: 5 °C min−1, ω = 1 rad s−1). | ||
3.3 Isothermal crystallization
δ at different frequencies was shown in Fig. 5 against the crystallization time. According to eqn (2), the critical state is characterized by a loss tangent which is frequency-independent. As seen in Fig. 5, the critical LST point was reached after 17261 s (4.8 h).
 | ||
| Fig. 4 Evolution of storage modulus (a) and loss modulus (b) at various angular frequencies during isothermal crystallization for M03 at 116 °C, obtained by cyclic frequency sweep. The scatter symbols are experimental data. The solid lines are fitting curves of logistic function. | ||
 | ||
| Fig. 5 Evolution of loss tangent during cyclic frequency sweeps for M03 at 116 °C. | ||
Because the experimental data at different frequencies in cyclic frequency sweep are not sampled at the same time, the interpolation procedures4,40,41 had been commonly adopted to obtain the data at a given crystallization time. As shown in Fig. 4, the experimental data were interpolated by using the logistic function. Then, frequency-sweep data of G′, G′′ and tanδ can be determined at any crystallization time, as shown in Fig. 6 and 7 for M series and L series, respectively. In accordance with the critical points determined from Fig. 5, the plots of tanδ versus frequency at the critical points exhibit the characteristic plateau of phase angle in the low frequency window (0.01–0.1 rad s−1), indicating the liquid–solid transition.
 | ||
| Fig. 6 Dependence of the dynamic moduli and loss tangent on frequency at different crystallization times for M01 at 122 °C (a) and M03 at 116 °C (b). The solid symbols and horizontal lines denote the critical state of liquid–solid transition. | ||
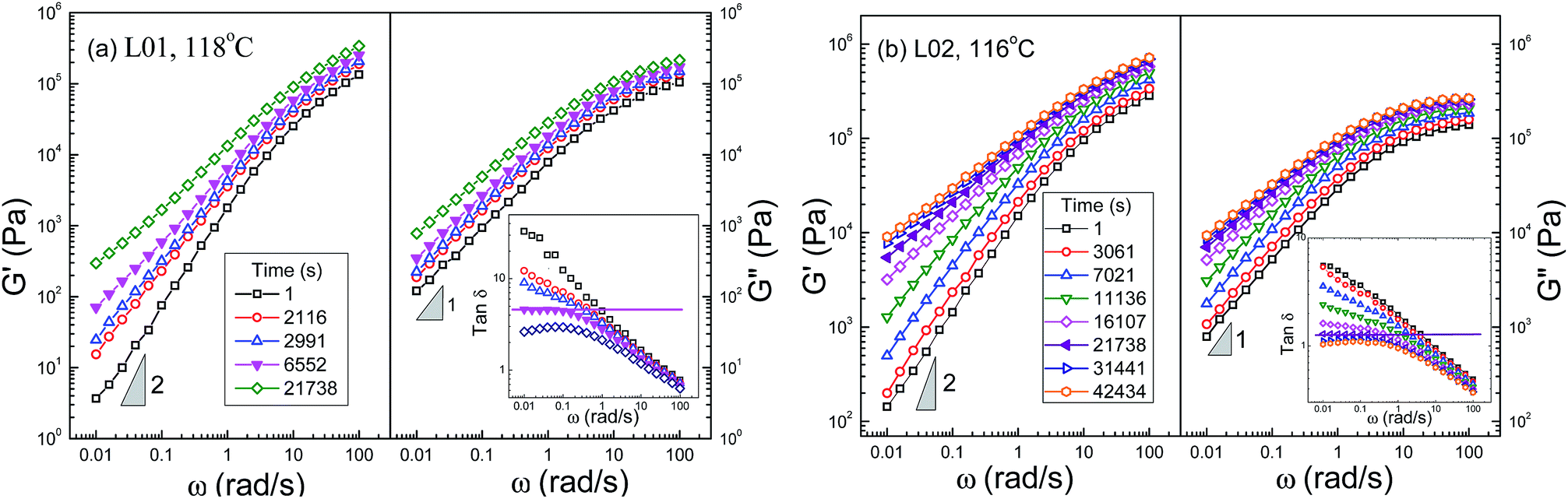 | ||
| Fig. 7 Dependence of the dynamic moduli and loss tangent on frequency at different crystallization times for L01 at 118 °C (a) and L02 at 116 °C (b). The solid symbols and horizontal lines denote the critical state of liquid–solid transition. | ||
One may argue if the LST from the frequency window 0.01–0.1 rad s−1 can reflect the true liquid–solid transition. Coppola et al.3 had found a long relaxation process for poly (1-butene) at a much lower frequency through the inverse quench method. However, in practice, it's still not clear to what extent the frequency is low enough to determine the longest relaxation process, reflecting the formation of spanning network at the largest length scale. On the other hand, the low frequency measurements require extremely long time to follow the crystallization process at very low undercoolings, and no applicable in practice. Due to these inherent problems, the frequency window of 0.01–0.1 rad s−1 had been generally adopted in various crystalline polymers4,5,13,29 to determine the critical state of LST. Since the present focus is the effect of mesophase separation on the formation of network at the same space-scale, the LST determined from the frequency window of 0.01–0.1 rad s−1 can be enough to give a direct comparison between different OBCs.
From Fig. 6 and 7, obvious differences of the frequency-sweep data can be found between the weakly segregated OBCs (M01, L01) and the strongly segregated OBCs (M03, L02). For M01, crystallization mainly affects G′, and has little influence on G′′. At the critical point, the storage moduli increase evidently at low frequencies, while show very weak increase at high frequencies. For M03, crystallization causes the increase of G′ and G′′ over a wide range of frequency from the beginning. Both G′ and G′′ increase greatly at the critical point. Similar phenomenon is also seen in L02, which is also strongly mesophase separated. The evolution of dynamic moduli of L01 during crystallization is similar to L02, but the enhancement in G′ and G′′ is much weaker. It suggests that the connecting structures of the network could be quite different among these OBCs, and strongly correlated to their mesophase separation.
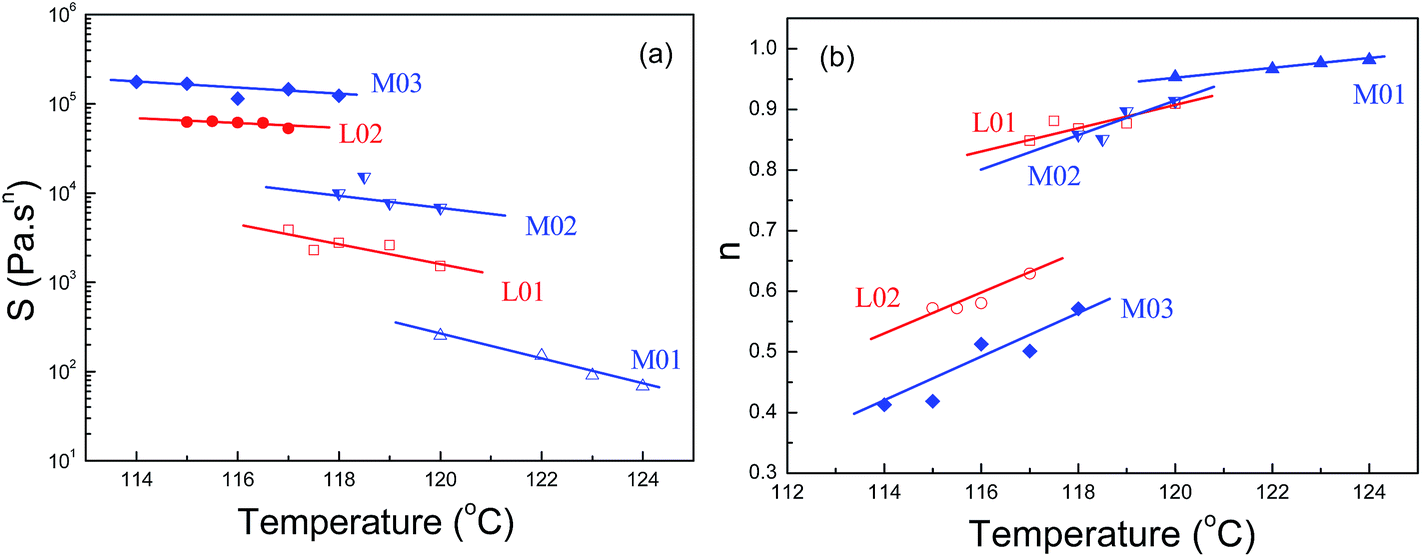 | ||
| Fig. 8 The gel stiffness (a) and relaxation exponent (b) at the critical states with the crystallization temperature for OBCs. Solid lines are linear regression fits. | ||
The critical properties are dependent on both crystallization temperature and Mw. These trends, however, are similar to those of homopolymers and random copolymer polymer.3,5,13 To isolate the contribution of mesophase separation on critical properties of OBCs, it is necessary to remove the influence of Mw on critical behaviors of homogeneous systems. Izuka et al.9 had investigated the chemical gelation process of polycaprolactone with different stoichiometric ratios, corresponding to different Mws. They found that when the logarithm of gel stiffness was plotted as a function of the relaxation exponent, all data with different Mws of chemical crosslinking polycaprolactone can be reduced to the same “universal” straight line, independently of either Mw. Similar phenomenon had been also found in physical gelation of homogeneous crystallizing polymers.3,5,13 Fig. 9 shows the critical gel stiffness plotted against the relaxation exponent for all OBCs at low undercoolings. Data from the work of Coppola and Acierno3,13 for homopolymer poly(1-butene) with different Mws, and data from the work of Horst et al.5 for a metallocene synthesized random copolymer of ethylene–butene containing 11 mol% 1-butene at various Tcs, were also included in Fig. 9 for comparison. As shown in Fig. 9, all data for poly(1-butene) with different Mws at various Tcs can fall on a single linear curve, suggesting that such plotting can eliminate the influence of Mw and Tc. They proposed the critical gel response is dictated by some intrinsic feature of the system, rather than by the specific physical conditions, such as the Mw or Tc, but the underlying mechanism had not been further illustrated.
 | ||
| Fig. 9 Gel stiffness as a function of the gel exponent at various crystallization temperatures for OBCs. Data from Coppola3 and Acierno13 for poly(1-butene) with different molecular weight, and data from Horst5 for ethylene–1-butene copolymer are included for comparison. The solid lines are linear regressions of the experimental data. | ||
In the chemical gelation process, the critical stiffness can be expressed as6,9,42
| S = G0λ0n | (3) |
S–n plot gives the characteristic relaxation time (logλ0) of the precursor molecule, and G0 is the plateau modulus of the fully cross-linked material. Then, the upper limit of S (n = 0) and the lower limit of S (n = 1) can be described by the plateau modulus (G0) and zero-shear viscosity (G0λ0) of the linear precursor, respectively. The slope of logS–n, i.e. the relaxation time of precursor, should be constant if the molecular weight of the precursor is fixed. However, it is interesting to find the linear relationship between logS and n in the crystallization of homopolymers, random copolymer as well as block copolymers (OBCs). Since different points in logS–n curve are determined at different crystallization temperatures, the apparent constant slope of logS–n implies the relaxation time (λ0) of chain segments between the physical crosslinking points are constant as the crystallization temperature varies, which implies that the length of chain segments must decrease as the crystallization temperature decreases.
Such presumption can be rationalized by the nucleation behavior during crystallization. Among the scenarios that suggested by Horst et al.,7 Acierno et al.8 and Coppola et al.,3 the direct contact between structural units are believed to be less possible. If the network is formed by the bridging molecules which have segments in neighboring crystalline phases, the relaxation time depends on the molecular weight of bridging segments (Ms). If the network is due to the impingement of amorphous chains, which are immobilized by their segments attachment within a crystalline structure, the relaxation time also depends on the molecular weight of amorphous segments (Ms). In either case, Ms is proportional to the distance between neighboring crystals. The mean nearest-neighbor distance l is proportional to the size of crystal d, and the ratio l/d depends only on the volume fraction of crystal and its geometry as well.43 The size of crystal can be expressed as
 | (4) |
| N ∝ Ṅ∝ e−Ea/kTe−ΔG/kT | (5) |
| Ms ∝ l ∝ d ∝ N−1/3 ∝ eEa/3kTeΔG/3kT | (6) |
The relaxation time λ0 depends on the molecular weight Ms through a power law,
| λ0 = kMsm | (7) |
| λ0 ∝ emEa/3kTemΔG/3kTe−Ea/kT | (8) |
It implies that the dependence of relaxation time on temperature could be very weak once the relationship mΔG = (3 − m)Ea is satisfied. Actually, Ea is usually independent of temperature for polyolefins, and the energy barrier ΔG depends on the undercooling ΔT by (ΔT)−2, whose change is also small in the limited temperature range for the rheological study of LST during crystallization. Therefore, there are two opposite effects on the relaxation of the critical network. The constant apparent relaxation time (linearity in logS–n) could be ascribed to the limited temperature range with low undercoolings. As the crystallization temperature decreases further, stronger dependence of the energy barrier ΔG on the undercooling ΔT may cause the failure of the linearity in logS–n at lower temperatures.
For a specific OBC, although the linear dependence of logS–n is seen, the Mw irrelevance of logS–n curve cannot be observed. As can be seen in Fig. 9, for OBC M-series, with the similar hard block content but varying Mw, the data points do not show the trend to fall on a single linear curve. In contrast, increasing the Mw from M01 to M02 and M03, the slope of logS–n decreases correspondingly, suggesting the increase of relaxation time of the critical network. The similar phenomenon is also seen in OBC L-series. As the logS–n plotting has eliminated the influence of Mw and Tc, the deviation of a linear relationship between different OBCs can be the contribution from the mesophase separation in OBC melts. Moreover, it is also found that the slopes of logS–n for L01 and M02 are very close. Since L01 and M02 have close molecular weight with similarly weak mesophase separation behavior, it implies that the characteristic relaxation time λ0 at the critical state does not rely on the hard block content, and only the extent of mesophase separation matters.
Furthermore, since the distance between physical crosslinking points is directly correlated with the characteristic relaxation time at the critical LST state, i.e., the slope of the logS–n plot, it can help to understand the different connecting structure of the network formed from different melt structures. From Fig. 9, a parallel relationship between the logS–n curves can be noted between the poly(1-butene) and ethylene–1-butene copolymer, indicating similar relaxation behaviors and connecting structures of the critical network in these homogeneous polymers. For the weakly segregated OBCs, the smaller relaxation time of the critical network implies the smaller distance between the crystallites, which act as the physical crosslinking points. In contrast, for the strongly segregated OBCs, the larger distance between the crystallites for the critical network can be inferred from the higher relaxation time. The different connecting structures of the crystallites at the critical points can be ascribed to the different crystallization mechanisms between the weakly segregated and the strongly segregated OBCs. It is also noticed that the slope of logS–n plot for M01 is even larger than that of homopolymer and random copolymer, indicating a smaller relaxation time in homogenous OBCs. It might be ascribed to the chain structure of the block copolymers. Compared to the homopolymer or random copolymer, the blocky OBC has a narrower block length distribution and much shorter average length of the amorphous blocks, which connect the neighboring crystals at the critical point of LST from the homogeneous melts. The shorter the average length of the amorphous segments in OBC M01, the faster the relaxation of the critical network, corresponding to the higher negative slope of logS–n.
The determination of the crystallinity by DSC at the critical point is difficult for OBCs because of the very weak heat flow change in isothermal experiments under low undercooling. In other polymer systems, alternative techniques had been adopted to evaluate the crystallinity at the critical point, which may give rise to different results as reported in different works.5,11 Here, we adopt the procedure employed in the work of Alizadeh et al.46 and Horst et al.,5 from which the absolute crystallinity can be calculated. At a given Tc, the isothermal crystallization by DSC was conducted for various time, then followed immediately a heating scan at a rate of 10 °C min−1 for each isothermal cycle. The typical heating traces with the time at a Tc are shown in Fig. 10 for OBCs M01 and M03. It can be seen that the melting peak shifts to higher temperatures and the peak area increases with the crystallization time. Considering the temperature range investigated by rheology, the above crystallization procedures by DSC were conducted under both large undercoolings and low undercoolings for a full comparison. Because of the slow crystallization rates under low undercoolings, the crystallization time would become extremely long to reach the completion of crystallization. So the maximum experimental time of the crystallization cycles was set to 360 min. Then, the corresponding heat of melting ΔHm can be plotted as a function of time at various temperatures, as shown in Fig. 11. By dividing the measured heat of melting by the theoretical value (ΔHm = 292 J g−1) of polyethylene with 100% crystallinity, the absolute crystallinity (Xc) of OBCs can be obtained at different stages of crystallization.
 | ||
| Fig. 10 Evolution of the melting behavior for OBCs after crystallization at Tc for various times for M01 at 122 °C (a) and M03 at 116 °C (b). The heating rate is 10 °C min. | ||
 | ||
| Fig. 11 Heat of melting with the crystallization time at various Tcs for OBCs M01 (a) and M03 (b). | ||
As shown in Fig. 11, at lower temperatures, a trend to approach a plateau of ΔHm in the late stage of crystallization can be found in M01 and M03. The star symbols in Fig. 11 denote the heat of melting at the critical points for M01 and M03 at higher crystallization temperatures. The critical values of ΔHm show weakly decrease with the increasing Tc, and are around 4.4 J g−1 and 14.0 J g−1 for M01 and M03, corresponding to the absolute crystallinity around 1.5% and 4.8%. The critical crystallinity for M01 is similar to the results of Horst et al.5 on a random copolymer ethylene–1-butene at various Tcs. But it is interesting to note that the critical crystallinity for M03 is much higher than that in M01. Such difference in crystallinity at the critical LST point for M01 and M03 further demonstrates that the OBC with strong mesophase separation has a different connecting structure at the critical point and should have a special underlying network formation mechanism as compared with that in homogeneous polymers.
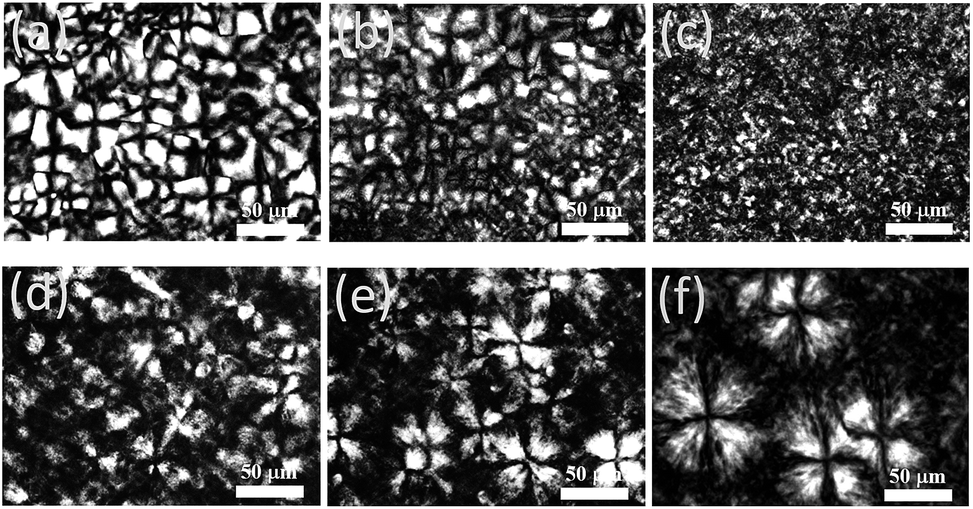 | ||
| Fig. 12 Polarized optical micrographs of OBCs as a function of the cooling rate: (a) M01 at 50 °C min, (b) M01 at 20 °C min, (c) M01 at 1 °C min, (d) M03 at 50 °C min, (e) M03 at 20 °C min−1, (f) M03 at 1 °C min−1. The scale bar is 50 μm. | ||
For homogeneous M01, the cooling rates have significant effects on the spherulite structures. As observed in Fig. 12a, large and well developed spherulites can be observed at large cooling rate, but decreasing the cooling rate appears to deteriorate the spherulite structure (Fig. 12b and c). Such cooling-rate dependent morphology, or more generally of the crystallization temperature regions dependent morphology, had been commonly found in homogeneous ethylene homopolymer,47,48 and also in ethylene–α-olefin random copolymer with low comonomer content.49 This can be described by the different regimes in the crystal growth theory proposed by Hoffman.48 For the crystallization conditions at slow cooling rate, or comparable to isothermal crystallization at higher Tc, called regime I crystallization, the deterioration of the spherulite structure cannot imply the lower degree of ordering for the crystalline structure. Conversely, since the nucleation rate is much slower than the lateral spreading rate of the nuclei at low undercoolings (regime I), the rod- or sheet-like crystalline structures with high degree of ordering are more preferred to be formed in regime I, leading to the poorly organized spherulite structures. At larger cooling rate, corresponding to the regimes II crystallization at lower Tc, the best developed spherulites are observed, but it does not indicate that the crystalline structure is more perfect. It can be explained that as the lateral growth rate of the nuclei is much slower than the surface nucleation rate at large undercoolings (regime II), the occurrence of the multiple nucleation on a monocystal layer leads to the formation of spherulite structures.
However, for strong mesophase separated M03, the opposite trend of morphology changes at various cooling rates from Fig. 12d–f can be observed. With the cooling rate decrease, the spherulites size become larger. It was found that the initial mesophase separation can promote the nucleation of crystallization,26,27 but fast mesophase separation kinetics had been found in M03,26 Thus, the extent of mesophase separation are little influenced by the cooling rate, in contrast to the great influence of cooling rate on phase separation kinetics of polymer blends.50 At slow cooling rates, the nucleation density is so low that limited numbers of crystals can breakout from the confinements of the microdomains. But once they breakout from the initial confinement, and they will gradually grow to larger spherulites because of the low nucleation density. However, at high cooling rates, the rapid cooling rate results in more nucleus appearing during cooling, thus, more crystals will breakout from the microdomains and continue to grow. No much more space is left for the spherulites to grow larger than that at slow cooling rates.
The spherulite growth during isothermal crystallization at high Tc are further shown in Fig. 13 for M01 and M03. Different morphology can be seen at the critical point of LST. For weakly segregated M01, large amount of tiny crystals are formed at the critical point (Fig. 13a). The system can be regarded as a physical network composed by the small crystalline regions connecting by polymer chains, which can go through amorphous and crystalline regions in the weakly segregated OBC. For strongly segregated M03, few large spherulites can be seen at the critical point (Fig. 13c). This can be explained that the initial crystal breakout is hindered by the mesophase separation, and the initial nucleation process is more similar to the sporadic nucleation in polypropylene. The system resembles a suspension of a solid phase dispersed in a liquid matrix. Liquid–solid transition happens at a high volume fraction of spherulite. The spherulite morphology in the late stage of crystallization can be found in Fig. 13b for M01 and Fig. 13d for M03. The morphologies of them at low undercooling agree well with the observation at slow cooling rate (Fig. 12).
 | ||
| Fig. 13 Polarized optical micrographs for M01 during isothermal crystallization at 121 °C at the critical LST point (tc = 3254 s) (a) and after crystallization for 12 hours (b), for M03 during isothermal crystallization at 116 °C at the critical LST point (tc = 17261 s) (c) and after crystallization for 12 hours (d). The scale bar is 50 μm. | ||
Fig. 14 presents a schematic illustration of the mechanism for understanding the effects of mesophase structures on the network formation of OBCs at the critical point of LST. In the molten state, the weakly segregated OBC (M01) shows a nearly disordered and mixing state of the hard segments and the soft segments, while the strongly segregated OBC (M03 and L02) can self-assemble into the microdomains in tens of nanometer. For weakly segregated OBC, when cooling from the melt, nucleation happens homogeneously in the sample and small amount of nanoscale crystals are sufficient to act as junctions linking together several neighboring chains to form a spanning network. The smaller crystals and then closer distance among crystals make the connecting chain segments shorter and relax faster. This is in consistent with the conclusions of the network connectivity in homogenous ethylene–butene copolymer5 with very low amount of crystallinity.
 | ||
| Fig. 14 Schematic illustration of the microstructures at the liquid–solid transitions of the melt weakly segregated OBC M01 and the melt strongly segregated OBC M03. The dash circles for the critical network indicate the rotational regions of the crystallites. | ||
However, in strongly segregated OBC, the initial formation and growth of crystals are strongly confined in the hard block rich microdomains. The network can start to form only after the crystals breakout the isolated microdomains, which is assisted by the short hard block dissolved in the soft blocks due to the polydispersity of OBCs. It explains the higher degree of crystallinity and the sparse large spherulites at the critical LST point of M03. Such retarded LST in OBCs during crystallization has not been observed in diblock copolymers. For strongly microphase separated diblock copolymers, the morphology of microphase separation in melt, with long-range order, can be maintained and stacks of lamellae are confined within the isolated domains.51–53 From the rheological observation, the modulus of such systems can increase after crystallization,54,55 but the transition from a liquid-like state to the solid-like state can hardly be observed. The critical network is hard to form in such systems since the crystals are confined in phase separated domains, and isolatedly distributed in the amorphous phase.
3.4 Critical point of LST: early stage or late stage of crystallization
It has been reported that the critical point appears at early stage of crystallization in homopolymers and random copolymers.3,5,30,33 Actually, rheological concept of liquid–solid transition has been suggested to study the early stage of crystallization of these polymers. However, we would argue that the critical point of LST might not happen at the early stage of crystallization especially for heterogeneous block copolymers with low overall crystallinity (7–14 wt%). To illustrate this, the evolutions of dynamic moduli with time for OBC M01 and M03 at a low frequency of 1 rad s−1 are presented in Fig. 15. At low temperatures, the dynamic rheological measurements were monitored through almost the whole crystallization process. As can be seen in Fig. 15, the initial increase of storage modulus for OBCs can be related to the crystallization induction period and nucleation period, and then the sharp increases in G′ correspond to crystal growth stage. In the end of the crystallization, G′ tend to increase gradually and can approach a plateau, which depends on the crystallinity and the crystal structure. Fig. 16 shows the master curves for the five OBCs during crystallization, which are obtained by shifting the isothermal plots at various crystallization temperatures (Tc) to a reference temperature, Tref, of 115 °C. The horizontal shift factor, aT, is determined by superimposing the storage modulus at different temperatures with that at Tref. The meaning of the horizontal shift factor had been found to be correlated to the crystallization rate constant consisted of the nucleation and growth rates.56,57 From Fig. 16a–c, the shifted isothermal curves can nearly overlap at various temperatures for M03, which has the highest molecular weight and the strongest mesophase separation among the M-series OBC with similar hard block content. As the molecular weight decrease from M03 to M02, and M01, the OBC in melts tend to become more homogenous, and more significant deviations of the isothermal plots from that at Tref can be seen in the late stage of crystallization. Similar phenomena can also be found for L01, L02, as shown in Fig. 16d and e. | ||
| Fig. 15 Evolution of storage modulus with time during isothermal crystallization for M01 (a) and M03 (b). The hollow symbols denote the time sweep data at 1 rad s−1, while the solid symbols denote the cyclic frequency data at 1 rad s−1. The dash lines denote the critical state of LST determined at low undercoolings. | ||
 | ||
| Fig. 16 The master curves for the isothermal crystallization of OBCs at a reference temperature of 115 °C. (a) M01, (b) M02, (c) M03, (d) L01, (e) L02. The dash lines denote the critical point of LST determined at low undercoolings. | ||
It is seen that the critical point of LST appears at very different stages in homogeneous OBC and heterogeneous OBC, indicated by the dash lines in Fig. 16. The critical point of LST appears at early stage of crystallization for M01, but intermediate stage for M03. Such trend is clearer in L-series OBC with lower crystallinity, i.e., the critical point of LST appears at late stage for L02. The delay of liquid–solid transition during crystallization in phase separated sample is strongly related to the confined effect of microdomains on the growth of crystals. In strongly mesophase separated OBCs, the isolated domains prevent the contact of crystals at the early stage, and the liquid–solid transition cannot happen before the crystals fully occupy and breakout the hard block rich domains. Therefore, it could be expected that liquid–solid transition at a Tc may possibly not happen if the hard block content is further decreased (as no enough crystallites to act as junctions) or phase separation is stronger in mesophase separated OBCs.
It should be pointed out that the critical point of LST cannot be determined for OBCs at large undercoolings, because the rapid crystallization rates at these Tc make it impossible to conduct the low frequency measurements. However, it is expected that the critical point of LST at large undercoolings could happen earlier in strongly mesophase separated OBCs. This can be ascribed to the promoted crystallization of short hard blocks that dissolved in the soft blocks from the beginning of crystallization. Thus, as Tc decreases, the occurrence of LST in strongly mesophase-separated OBCs would be more close to that in homogeneous systems.
To sum up, two factors can be important for LST. First, the overall crystallinity should be above a critical value for a sample. For the LST at a specific crystallization temperature, a minimum amount of crystallinity is required to act as junctions to form the critical network. Second, the aggregation structure of the crystals at LST can be the other factor determining the structure of the critical network at LST, which is influenced by the mesophase separation in melts. For the weakly segregated OBCs, the random crystallization from the nearly homogenous melts makes it possible to form a critical network at a very low crystallinity in the earlier stage of crystallization. For the strongly segregated OBCs, as the strong confinement of the mesophase separated microdomains, the critical network can be formed only after the crystals breakout the isolated microdomains, corresponding to the higher crystallinity in the later stage of crystallization. Therefore, the formation of the critical network in OBCs with different melt structures are the results of the different aggregation structure of the spherulites at LST.
4. Conclusions
In this work, the effect of mesophase separation on the evolution of viscoelastic properties during crystallization had been studied in a series multiblock OBCs with different molecular structures. The liquid–solid transition was monitored by the cyclic frequency sweep at low undercoolings to understand the network formation mechanism at critical transition point for OBCs with different melt structures. It is found that the formation of critical network during LST is postponed by the mesophase separated domains. As compared to weakly segregated OBCs, the critical point of LST in strongly segregated OBCs happens at intermediate or even late stage of crystallization with much higher crystallinity and large spherulites. Such delayed transition is caused by the confining effect of mesophase separated microdomains on the growth of crystals, which need to breakout the microdomains to form network. Such observation indicates that the solidification process due to crystallization in heterogeneous block copolymers can be very different from that in homogeneous polymers.Acknowledgements
This work is supported by National Science Foundation of China (21474063) and the National Basic Research Program of China (2011CB606005). W. Yu is supported by the Program for New Century Excellent Talents in University and the SMC project of Shanghai Jiao Tong University.Notes and references
- Z. Ma, L. Balzano, G. Portale and G. W. M. Peters, Polymer, 2014, 55, 6140–6151 CrossRef CAS PubMed.
- N. Tian, D. Liu, L. Meng, W. Zhou, T. Hu, X. Li and L. Li, RSC Adv., 2014, 4, 9632–9638 RSC.
- S. Coppola, S. Acierno, N. Grizzuti and D. Vlassopoulos, Macromolecules, 2006, 39, 1507–1514 CrossRef CAS.
- N. V. Pogodina and H. H. Winter, Macromolecules, 1998, 31, 8164–8172 CrossRef CAS.
- R. H. Horst and H. H. Winter, Macromolecules, 2000, 33, 130–136 CrossRef CAS.
- H. H. Winter and M. Mours, Adv. Polym. Sci., 1997, 134, 165–234 CrossRef CAS.
- Y. Wang, A. Lue and L. Zhang, Polymer, 2009, 50, 5474–5481 CrossRef CAS PubMed.
- J. Wu, Z. L. Wu, H. Yang and Q. Zheng, RSC Adv., 2014, 4, 44030–44038 RSC.
- A. Izuka, H. H. Winter and T. Hashimoto, Macromolecules, 1992, 25, 2422–2428 CrossRef CAS.
- A. Elmoumni, H. H. Winter, A. J. Waddon and H. Fruitwala, Macromolecules, 2003, 36, 6453–6461 CrossRef CAS.
- Y. Gelfer and H. Winter, Macromolecules, 1999, 32, 8974–8981 CrossRef.
- N. V. Pogodina, V. P. Lavrenko, S. Srinivas and H. H. Winter, Polymer, 2001, 42, 9031–9043 CrossRef CAS.
- S. Acierno, N. Grizzuti and H. H. Winter, Macromolecules, 2002, 35, 5043–5048 CrossRef CAS.
- D. J. Arriola, E. M. Carnahan, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, Science, 2006, 312, 714–719 CrossRef CAS PubMed.
- F. Zuo, C. Burger, X. M. Chen, Y. M. Mao, B. S. Hsiao, H. Y. Chen, G. R. Marchand, S. Y. Lai and D. Chiu, Macromolecules, 2010, 43, 1922–1929 CrossRef CAS.
- S. Wu, H. Li, G. Huang and J. Wu, RSC Adv., 2014, 4, 19024–19033 RSC.
- Z.-Z. Tong, B. Zhou, J. Huang, J.-T. Xu and Z.-Q. Fan, RSC Adv., 2014, 4, 15678–15688 RSC.
- Z. Li, Y. Shi, H. Liu, F. Chen, Q. Zhang, K. Wang and Q. Fu, RSC Adv., 2014, 4, 45234–45243 RSC.
- H. P. Wang, D. U. Khariwala, W. Cheung, S. P. Chum, A. Hiltner and E. Baer, Macromolecules, 2007, 40, 2852–2862 CrossRef CAS.
- D. U. Khariwala, A. Taha, S. P. Chum, A. Hiltner and E. Baer, Polymer, 2008, 49, 1365–1375 CrossRef CAS PubMed.
- J. Jin, J. Du, Q. Xia, Y. Liang and C. C. Han, Macromolecules, 2010, 43, 10554–10559 CrossRef CAS.
- S. Li, R. A. Register, B. G. Landes, P. D. Hustad and J. D. Weinhold, Macromolecules, 2010, 43, 4761–4770 CrossRef CAS.
- G. Liu, Y. Guan, T. Wen, X. Wang, X. Zhang, D. Wang, X. Li, J. Loos, H. Chen, K. Walton and G. Marchand, Polymer, 2011, 52, 5221–5230 CrossRef CAS PubMed.
- S. Li, R. A. Register, J. D. Weinhold and B. G. Landes, Macromolecules, 2012, 45, 5773–5781 CrossRef CAS.
- T. Wen, G. Liu, Y. Zhou, X. Zhang, F. Wang, H. Chen, J. Loos and D. Wang, Macromolecules, 2012, 45, 5979–5985 CrossRef CAS.
- P. He, W. Shen, W. Yu and C. Zhou, Macromolecules, 2014, 47, 807–820 CrossRef CAS.
- Z. Z. Tong, B. Zhou, J. Huang, J. T. Xu and Z. Q. Fan, Macromolecules, 2014, 47, 333–346 CrossRef CAS.
- S. Cossar, D. Nichetti and N. Grizzuti, J. Rheol., 2004, 48, 691–703 CrossRef CAS.
- M. Gelfer, R. H. Horst, H. H. Winter, A. M. Heintz and S. L. Hsu, Polymer, 2003, 44, 2363–2371 CrossRef CAS.
- R. H. Horst and H. H. Winter, Macromolecules, 2000, 33, 7538–7543 CrossRef CAS.
- Y. G. Lin and H. H. Winter, Macromolecules, 1988, 21, 2439–2443 CrossRef CAS.
- D. Nichetti, S. Cossar and N. Grizzuti, J. Rheol., 2005, 49, 1361–1376 CrossRef CAS.
- N. V. Pogodina, S. K. Siddiquee, J. W. van Egmond and H. H. Winter, Macromolecules, 1999, 32, 1167–1174 CrossRef CAS.
- H. W. Richtering, K. D. Gagnon, R. W. Lenz, R. C. Fuller and H. H. Winter, Macromolecules, 1992, 25, 2429–2433 CrossRef CAS.
- M. van Gurp and J. Palmen, Rheol. Bull., 1998, 67, 5–8 Search PubMed.
- C. X. Zhou, R. M. Li and W. Yu, J. Macromol. Sci., Part B: Phys., 2006, 45, 889–898 CrossRef PubMed.
- P. H. P. Macaúbas and N. R. Demarquette, Polym. Eng. Sci., 2002, 42, 1509–1519 Search PubMed.
- J. D. Ferry, Viscoelastic properties of polymers, John Wiley & Sons, New York, 3rd edn, 1980 Search PubMed.
- R. G. Alamo, B. D. Viers and L. Mandelkern, Macromolecules, 1993, 26, 5740–5747 CrossRef CAS.
- M. Mours and H. H. Winter, Rheol. Acta, 1994, 33, 385–397 CrossRef CAS.
- K. Te Nijenhuis and H. H. Winter, Macromolecules, 1989, 22, 411–414 CrossRef CAS.
- J. C. Scanlan and H. H. Winter, Macromolecules, 1991, 24, 47–54 CrossRef CAS.
- S. Torquato, B. Lu and J. Rubinstein, Phys. Rev. A, 1990, 41, 2059–2075 CrossRef CAS.
- L. Mandelkern, Crystallization of polymers, McGraw-Hill, New York, 1964 Search PubMed.
- C. Schwittay, M. Mours and H. H. Winter, Faraday Discuss., 1995, 101, 93–104 RSC.
- A. Alizadeh, L. Richardson, J. Xu, S. McCartney, H. Marand, Y. W. Cheung and S. Chum, Macromolecules, 1999, 32, 6221–6235 CrossRef CAS.
- L. Mandelkern, M. Glotin and R. A. Benson, Macromolecules, 1981, 14, 22–34 CrossRef CAS.
- J. D. Hoffman, L. J. Frolen, G. S. Ross and J. I. Lauritzen Jr, J. Res. Natl. Bur. Stand., Sect. A, 1975, 79, 671–699 CrossRef.
- M. Peeters, B. Goderis, C. Vonk, H. Reynaers and V. Mathot, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 2689–2713 CrossRef CAS.
- D. Dutta, H. Fruitwala, A. Kohli and R. A. Weiss, Polym. Eng. Sci., 1990, 30, 1005–1018 CAS.
- D. J. Quiram, R. A. Register and G. R. Marchand, Macromolecules, 1997, 30, 4551–4558 CrossRef CAS.
- T. Shiomi, H. Tsukada, H. Takeshita, K. Takenaka and Y. Tezuka, Polymer, 2001, 42, 4997–5004 CrossRef CAS.
- Y.-L. Loo, R. A. Register and A. J. Ryan, Macromolecules, 2002, 35, 2365–2374 CrossRef CAS.
- S. Nojima, D. Inokawa, T. Kawamura and K. H. Nitta, Polym. J., 2008, 40, 986–991 CrossRef CAS.
- A. Kelarakis, S. M. Mai, C. Booth and A. J. Ryan, Polymer, 2005, 46, 2739–2747 CrossRef CAS PubMed.
- D. Lellinger, G. Floudas and I. Alig, Polymer, 2003, 44, 5759–5769 CrossRef CAS.
- T. V. Chan, G. D. Shyu and A. I. Isayev, Polym. Eng. Sci., 1995, 35, 733–740 CAS.
| This journal is © The Royal Society of Chemistry 2015 |