
Structure–inhibition relationship of phenylethanoid glycosides on angiotensin-converting enzyme using ultra-performance liquid chromatography-tandem quadrupole mass spectrometry†
Abstract
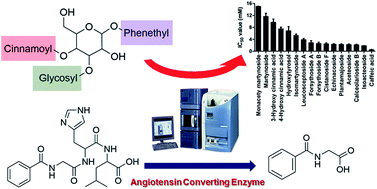
Angiotensin-converting enzyme (ACE) plays a critical role in the rennin–angiotensin system. Recently, natural products isolated from herbal medicines have demonstrated an inhibitory effect against ACE, suggesting their potential value in the regulation of blood pressure. In the present study, the ACE inhibition (ACEI) provided by 21 phenylethanoid glycosides and related phenolic compounds was investigated by measuring the production of hippuric acid (HA) using a rapid, sensitive, accurate and specific ultra-performance liquid chromatography-tandem quadrupole mass spectrometry (UPLC-MS/MS) method. The test compounds showed a wide spectrum of inhibitory potency on ACE at 50 mM, ranging from 5.29 to 95.01%, and compounds with ACEI greater than 50% were selected for IC50 determination, a measure of the effectiveness of a substance in inhibiting a specific biological function. The IC50 values ranged from 0.53 ± 0.04 to 15.035 ± 0.036 mM. The structure–inhibition relationships were then explored and showed that cinnamoyl groups played an essential role in the ACEI of phenylethanoid glycosides. Furthermore, the substructure of increasing ACEI in phenylethanoid glycosides involved a greater number of hydroxyl groups and less steric hindrance, allowing chelation by the active Zn2+ site of the ACE. Our results confirmed that phenylethanoid glycosides offer a widely available source of anti-hypertensive natural products. In addition, the information provided by the study of structure–inhibition relationships will assist in the further design of structurally modified phenylethanoid glycosides as anti-hypertensive drugs.
 Please wait while we load your content...
Please wait while we load your content...

