
Synthesis and evaluation of new benzimidazole-based COX inhibitors: a naproxen-like interaction detected by STD-NMR†
Abstract
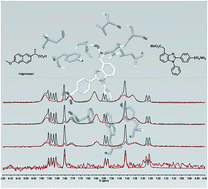
Non-steroidal anti-inflammatory drugs exert their pharmacological activity through inhibition of cyclooxygenase 1 and 2 (COX-1 and COX-2). Recent research suggests that a balanced inhibition of both COX-1 and COX-2 is the key to reduce the side-effects exhibited by COX inhibitors. We developed new benzimidazole-based compounds that showed a balanced COX inhibition, supported by molecular docking screening. The human whole blood assays demonstrated that the ester derivatives were potent inhibitors. Competitive saturation transfer difference (STD)-NMR experiments, in the presence of COX-2, using naproxen and diclofenac demonstrated that ester derivatives do not compete with diclofenac for the same binding site, but compete with the allosteric inhibitor naproxen. Combination of NMR spectroscopy with molecular docking has permitted us to detect a new naproxen-like inhibitor, which could be used for future drug development.
 Please wait while we load your content...
Please wait while we load your content...

