
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tropospheric chemical degradation of vinyl and allyl acetate initiated by Cl atoms under high and low NOx conditions†
María B.
Blanco
a,
Iustinian
Bejan
bc,
Ian
Barnes
*b,
Peter
Wiesen
b and
Mariano A.
Teruel
*a
aInstituto de Investigaciones en Fisicoquímica de Córdoba (I.N.F.I.Q.C.), Dpto. de Fisicoquímica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba. Ciudad Universitaria, 5000 Córdoba, Argentina. E-mail: mteruel@fcq.unc.edu.ar; Fax: +54 351 4334188; Tel: +54 351 5353866 int. 53531
bPhysikalische & Theoretische Chemie/FBC, Bergische Universitaet Wuppertal, Wuppertal, Germany. E-mail: barnes@uni-wuppertal.de; Fax: +49 202 439 2505; Tel: +49 202 439 2510
c“Al. I. Cuza” University of Iasi, Faculty of Inorganic and Analytical Chemistry, Iasi, Romania
First published on 22nd May 2015
Abstract
The products of the reactions of Cl atoms with vinyl acetate (VA) and allyl acetate (AA) have been investigated in a 1080 L chamber using in situ FTIR. The experiments were performed at 296 K and atmospheric pressure of synthetic air in the presence and in the absence of NOx. For the reaction of Cl with VA in the presence of NOx formic acetic anhydride, acetic acid and formyl chloride are the major reaction products. In the absence of NOx, the yields of these products are significantly reduced and formation of the carbon-chain-retaining compound CH3C(O)OC(O)CH2Cl is observed. For the reaction of Cl with AA in the presence of NOx acetoxyacetaldehyde and formaldehyde were observed as the main products. In contrast, without NOx, the observations support that the major reaction pathway is the formation of the carbon-chain-retaining compound CH3C(O)OCH2C(O)CH2Cl. The reaction mechanisms leading to the products are discussed. The formation of the high yields of formyl chloride and formaldehyde in the reactions of Cl with VA and AA, respectively, are at odds with currently accepted mechanistic pathways.
Introduction
In addition to emissions from natural sources (vegetation and biomass combustion) industrial activities have resulted in the wide-spread release of acetates to the atmosphere.1 Emissions from automobiles represent another source of these compounds; a number of acetates, including vinyl acetate (VA: CH3C(O)OCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) and allyl acetate (AA: CH3C(O)OCH2CHCH2), have been detected as products in the combustion of rape methyl esters used as fuel alternatives or additives.2
CH2) and allyl acetate (AA: CH3C(O)OCH2CHCH2), have been detected as products in the combustion of rape methyl esters used as fuel alternatives or additives.2
The gas-phase removal processes of these esters from the atmosphere, and volatile organic compounds (VOCs) in general, is mainly through their reactions with OH, NO3, and O3 with removal by Cl being thought to play, under most circumstances, a very minor role since the global Cl atom concentration is low (<103 radicals cm−3).3–5 The general conception has been that any significant removal of VOCs through reaction with Cl atoms is constrained to (i) the marine boundary layer and coastal regions,6,7 and (ii) industrial areas where chlorine can be emitted directly from incineration and power generation.8,9 However, a series of recent observations of nitryl chloride (ClNO2) in marine and mid-continental air10,11 and in urban power plants plumes12 have highlighted the potential importance of ClNO2 as a hither to unconsidered source of Cl and indicated that the role of Cl atoms in initiating the oxidation of VOCs may be more widespread and significant than previously thought. Nitryl chloride is formed during the night via the heterogeneous reaction of N2O5 with particle Cl− and photolyzes readily in the early morning to produce Cl and an NO2 molecule.13 A field and modelling study of Cl2 and ClNO2 in the coastal marine boundary layer has recently been reported for the Los Angeles region.14 The results indicated that over the course of an entire model day ClNO2 accounted for 45% of the integrated Cl-atom production compared to only 10% for Cl2 with the remaining 45% being attributed to the reaction of OH with HCl. The study also showed that Cl-atom mediated chemistry could contribute quite substantially to the photooxidation of VOCs in this region particularly in the early morning.
In order, to evaluate the contribution of VA and AA to the oxidative capacity of the atmosphere and potential environmental and health effects detailed kinetic and mechanistic information on their tropospheric gas-phase degradation pathways are required. There have been several kinetic studies of the reactions of VA and AA with OH,15–17 NO3,17 O3 (ref. 17) and Cl.18 In addition, product studies on the OH-radical initiated oxidation of these unsaturated esters have also been reported. Picquet-Varrault et al. have reported product studies on the reactions of OH radicals with both VA17 and AA19 performed at atmospheric pressure and room temperature in the presence of NOx using FTIR spectrometry for the analyses. Picquet-Varrault et al.17 reported formaldehyde, formic acetic anhydride and acetic acid as the main products formed in the reaction of OH with VA. For the reaction of OH with AA acetoxyacetaldehyde and HCHO were the main products observed.19
We have recently reported room temperature product investigations on the gas-phase reactions of OH radicals with VA and AA in the presence and absence of NOx.20 Our results in the presence of NOx agreed well with those reported by Picquet-Varrault et al.17–19 for the two compounds. However, our studies showed that in the absence of NOx there are significant changes in the relative importance of the possible product channels for the reaction of the 1,2-hydroxyalkoxy radicals formed after addition of OH to the double bond in the compounds. In the case of VA, an α-ester rearrangement to produce CH3C(O)OH and CH2(OH)CO˙ radicals rather than decomposition to form formic acetic anhydride dominated. In the case of AA reaction of the 1,2-hydroxyalkoxy radical with O2 to form the carbon chain-retaining product 3-hydroxy-2-oxo-propyl ester (CH3C(O)OCH2C(O)CH2OH) dominated in the absence of NOx contrasting very sharply with the decomposition channel of the 1,2-hydroxyalkoxy radicals to form acetoxyacetaldehyde and HCHO, which dominated when the reaction was studied in the presence of NOx.
In a continuation of our investigations on the photooxidation mechanisms of unsaturated VOCs for different NOx scenarios, we report here product studies on the Cl-atom initiated photooxidation of VA and AA in the presence and absence of NOx:
| Cl + CH2CHOC(O)CH3 (VA) → products | (1) |
| Cl + CH2CHCH2OC(O)CH3 (AA) → products | (2) |
A computational study on the mechanism and kinetics of the Cl-initiated oxidation of VA in the presence of NOx was reported recently.21 The study found that Cl addition to the double bond is the dominate pathway followed, in the presence of O2 and NO, by decomposition to give the major products observed in our work.
To the best of our knowledge this is the first reported experimental product investigation of the Cl-atom initiated photooxidation for both VA and AA performed in the presence and in the absence of NOx.
Experimental section
All the experiments were performed in a 1080 L quartz-glass reaction chamber at (298 ± 2) K and a total pressure of (760 ± 10) Torr of synthetic air (760 Torr = 101.325 kPa). A detailed description of the reactor can be found elsewhere22 and only a brief description is given here. The photolysis system consists of 32 superactinic fluorescent lamps (Philips TL05 40W: 320–480 nm, λmax = 360 nm), which are spaced evenly around the reaction vessel. The chamber is equipped with a White type multiple-reflection mirror system with a base length of (5.91 ± 0.01) m for sensitive in situ long path infrared absorption monitoring of reactants and products in the spectral range 4000–700 cm−1. The White system was operated at 82 traverses, giving a total optical path length of (484.7 ± 0.8) m. Infrared spectra were recorded with a spectral resolution of 1 cm−1 using a Nicolet Nexus FT-IR spectrometer equipped with a liquid nitrogen cooled mercury-cadmium-telluride (MCT) detector.Chlorine atoms were generated by the photolysis of Cl2 using the fluorescent lamps:
| Cl2 + hν → 2Cl | (3) |
Experiments were performed on mixtures of Cl2/acetate/air or Cl2/acetate/NO/air which were irradiated for periods up to 5 minutes during the course of which infrared spectra were recorded at 1 cm−1 resolution with the FTIR spectrometer. Tests showed that photolytic removal of the acetates in the absence of Cl2 and NO was negligible. The systems, in particular the AA system, are very reactive and typically 20 interferograms were co-added per spectrum over a period of approximately 5 min for the experiments with VA and 10 interferograms for the experiments with AA. The concentrations of the reactants used in the experiments and the infrared frequencies used for monitoring are given in the ESI.†
The quantification of reactants and products was performed by comparison with calibrated reference spectra contained in the IR spectral data bases of the laboratories in Wuppertal and Laboratoire Interuniversitaire des Systemes Atmosphériques (LISA), Paris, France.23
Results
Cl + vinyl acetate
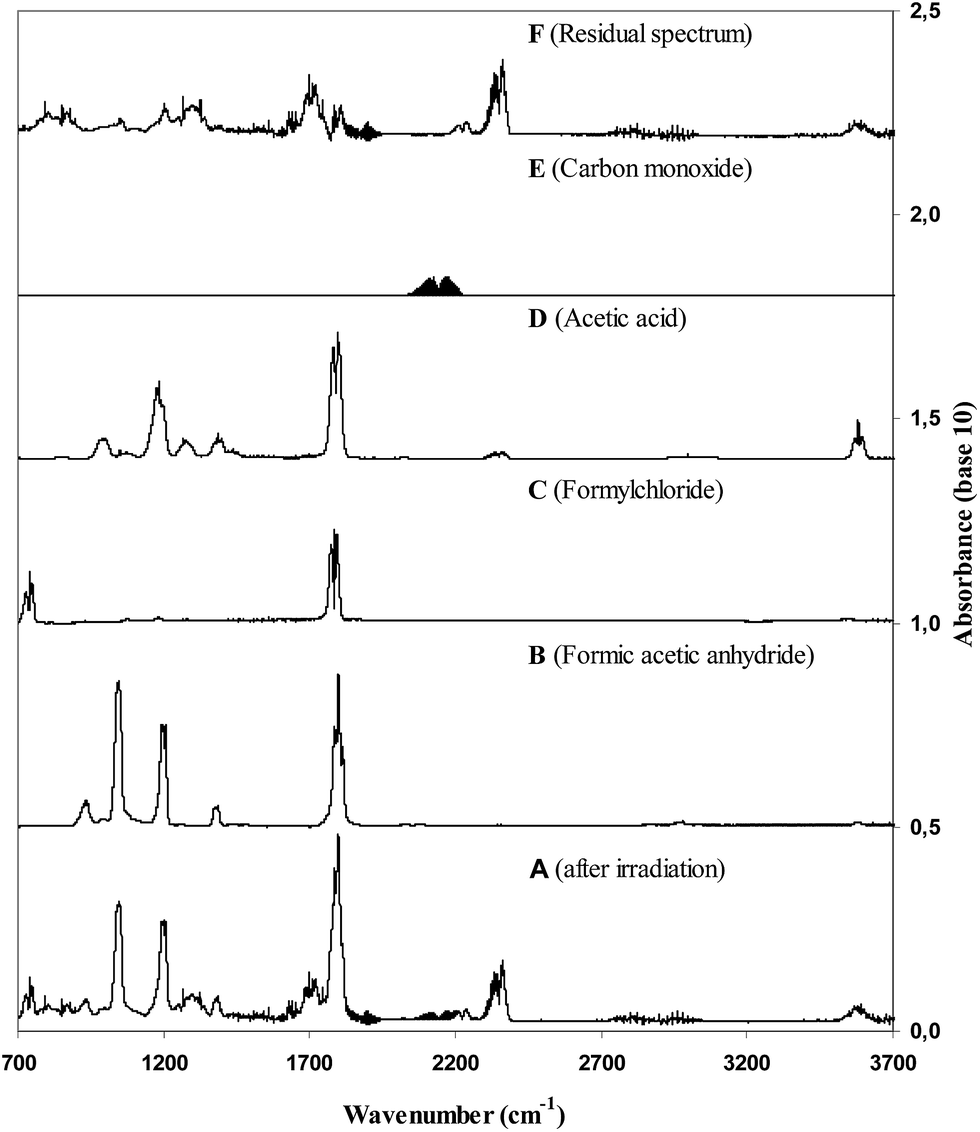 | ||
| Fig. 1 Panel A shows the product spectrum obtained from the irradiation of a VA/Cl2/NO/air reaction mixture. Panels B, C. D and E show reference spectra of formic acetic anhydride, formyl chloride, acetic acid, and carbon monoxide, respectively. Panel F shows the residual product spectrum obtained after the subtraction of the identified products. | ||
Concentration–time profiles of VA and the identified products, formic acetic anhydride, formyl chloride, acetic acid and carbon monoxide are shown in Fig. S1 of the ESI.† Plots of the concentrations of the identified products as a function of reacted VA are shown in Fig. S2.† As can be seen in Fig. S1† the increase in the product concentrations scale linearly with the loss of VA supporting that the products are all primary and that secondary loss or formation of these products is insignificant. The molar yields of the products observed in the reaction of Cl with VA in the presence of NOx, obtained from the slopes of the plots in Fig. S2,† are listed in Table 1.
| Acetate | Product | Yield (%) (NOx – containing) | Yield (%) (NOx – free) |
|---|---|---|---|
| a Lower limit observations; attempted corrections for secondary consumption support a much higher yield (see text). b Estimate based on experimental observations. | |||
| CH3C(O)OCHCH2 (vinyl acetate) |
CH3C(O)OC(O)H (formic acetic anhydride) | 69 ± 8 | 25 ± 5 |
| CH3C(O)OC(O)CH2Cl | — | ∼50a | |
| CH3C(O)OH | 27 ± 5 | 12 ± 3 | |
| HC(O)Cl | 94 ± 12 | 48 ± 8 | |
| HC(O)H | ≤5 | — | |
| CO | 30 ± 5 | 15 ± 4 | |
| CH3C(O)OCH2CHCH2 (allyl acetate) |
CH3C(O)OCH2C(O)H (acetoxyacetaldehyde) | 66 ± 8a | ≤5 |
| CH3C(O)OCH2C(O)CH2Cl | ≤5 | ≥95b | |
| CH3C(O)OC(O)H (formic acetic anhydride) | Secondary | ≤5 | |
| CH3C(O)OH | Secondary | ≤5 | |
| HC(O)Cl | Secondary | ≤5 | |
| HC(O)H | >90 | ≤5 | |
| CO | Secondary | ≤5 | |
Many of the absorption features in the residual product spectrum correlate very well with those of acetic acid anhydride and chloroacetone indicating that the compound (or compounds) giving rise to the IR features contains structural elements common to both acetic acid anhydride and chloroacetone. Since we are certain that all other possible reaction products from the reaction of Cl with VA have been accounted for we are confident that the unidentified absorptions are mainly due to the formation of the VA carbon-skeleton retaining compound chloro-acetic acid anhydride (CH3C(O)OC(O)CH2Cl) although contributions from CH3C(O)OC(OH)CH2Cl can not be excluded. Further justification for the formation of CH3C(O)OC(O)CH2Cl from the 1,2-chloroalkoxy radicals formed in the Cl + vinyl acetate reaction are given in the discussion on the formation of CH3C(O)OCH2C(O)CH2Cl in the reaction of Cl with allyl acetate.
Concentration–time profiles of VA and the four identified products are shown in the ESI in Fig. S4.† Plots of the concentrations of the products as a function of reacted VA for the NOx-free reaction system are shown in Fig. 2. The plots are linear with near zero intercepts supporting that the products are all primary. The molar yields of the products observed in the reaction of Cl with VA in the absence of NOx, obtained from the slopes of the plots in Fig. 2, are listed in Table 1.
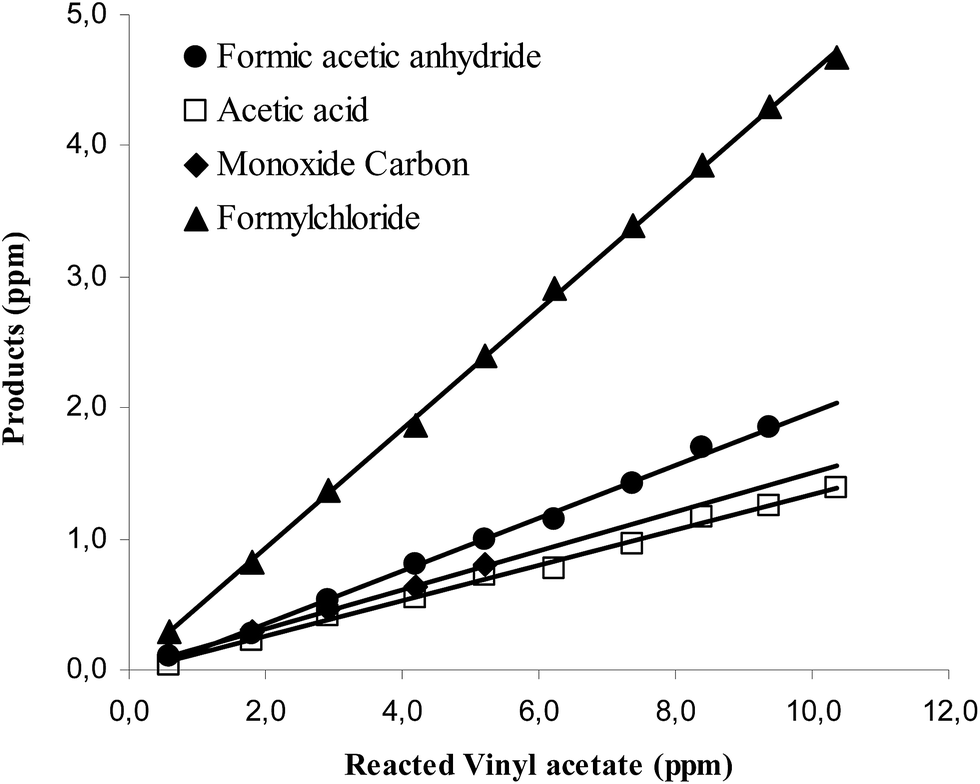 | ||
| Fig. 2 Plot of the concentrations of the products formed in the reaction of Cl with VA in the absence of NO as a function of the amount of reacted VA. | ||
Cl + allyl acetate
Fig. S6† shows the concentration–time profiles of AA and the identified products formed in the NOx-containing system. The profile of AA shows that it is being consumed very rapidly in the system. The initial concentration–time profiles of acetoxyacetaldehyde and HCHO are similar and indicate that they are both primary products. However, secondary removal of acetoxyacetaldehyde and especially HCHO in secondary reactions with Cl is clearly evident.
Although the concentration–time profiles of acetic acid, formic acetic anhydride and CO look as though they may be primary products, plots of their concentrations against the amount of AA consumed have non-zero intercepts and upward curvature which increases with increasing reaction time (see Fig. S7 in the ESI†). This behavior is indicative of formation of these compounds in secondary reactions. The formation of acetic acid and formic acetic anhydride in the system is attributed to fast secondary reactions of acetoxyacetaldehyde with Cl (see Discussion). Formation of formyl chloride was also observed in the system; however, it was only observed in small quantities toward the end of the reaction and is obviously being formed in secondary reactions.
The initial slopes of the plots of the concentrations of acetoxyacetaldehyde and HCHO against reacted AA indicate high yields for both compounds. Taking the first few points of a plot of the concentration of acetoxyacetaldehyde as a function of reacted AA gives a yield of acetoxyacetaldehyde of close to unity, however, if all the points are taken a yield of around 66% is obtained caused by curvature in the plot from fast secondary reaction of acetoxyacetaldehyde with Cl. Correction of yield for formaldehyde for secondary consumption with Cl using the method outlined in Tuazon et al.24 and a rate coefficient of 7.32 × 10−11 cm3 molecule−1 s−1 for the reaction of Cl with HCHO25 gives a yield of >90% for HCHO. The rate coefficient for the reaction of Cl with acetoxyacetaldehyde is not known. Using the rate coefficient for Cl with HCHO for the correction gave a yield of around 120%. Since the rate coefficient for Cl with acetoxyacetaldehyde is not likely to be much less than that for Cl with HCHO this would support a yield much higher than 66% yield for acetoxyacetaldehyde.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 fluorotelomer acrylate (C4F9CH2CH2OC(O)CHCH2) both in the presence and absence of NOx.
:2 fluorotelomer acrylate (C4F9CH2CH2OC(O)CHCH2) both in the presence and absence of NOx.
Further support comes from recent work on long chain Cx to C9 terminal alkenes by Walavalkar et al.28 which showed that addition of Cl to the inner carbon of the double bond is minimal and that 1-chloro-2-ketones are the main products in the absence of NOx. The above points serve to strengthen the arguments offered for the identification of acetic acid 3-chloro-2-oxo-propyl ester as the main product in the reaction of Cl with ally acetate in the absence of NO.
The near quantitative formation of acetic acid 3-chloro-2-oxo-propyl ester in the reaction of Cl with AA in the absence of NOx allows an approximate IR calibration of the compound. Using the band at 3415 cm−1 which is unique to the compound allows a yield of ≤5% to be placed on the formation of the ester in the reaction of Cl with AA in the presence of NOx.
Discussion
Cl + vinyl acetate
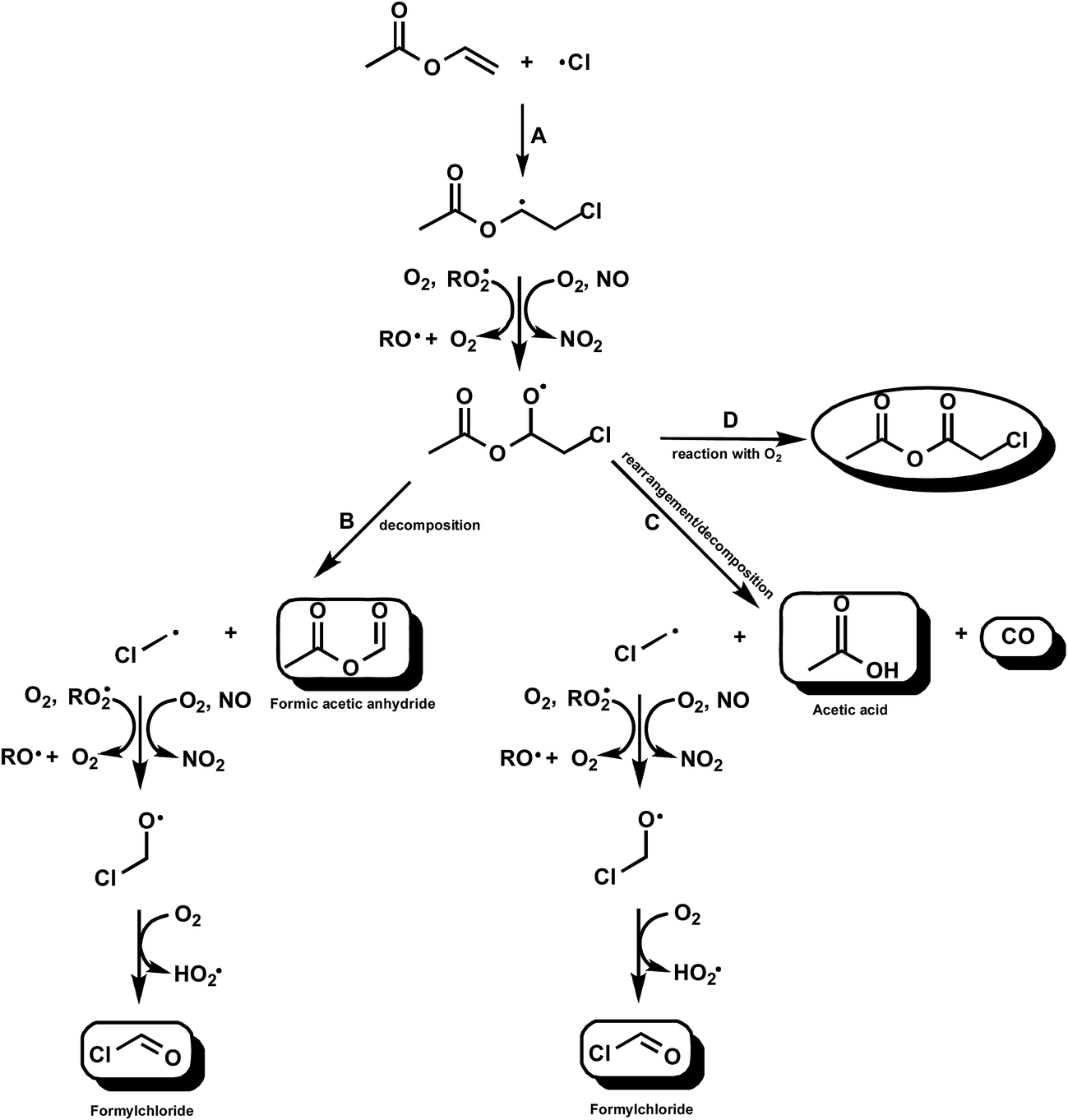 | ||
| Fig. 3 Simplified mechanism for the Cl-atom initiated oxidation of VA (CH2CHOC(O)CH3) via addition of Cl to the terminal carbon of the double bond. | ||
However, Bilde et al.29 have studied the reaction of Cl–CH2˙ radicals in N2 with different partial pressures of O2 in the presence and absence of NOx. They produced the radicals via reaction of Cl with CH3Cl. They did not measure the decay of CH3Cl since this was too small and determined the product yields from the sum of the three identified carbon-containing products HCOCl, HCHO and CO which they assumed was equal the amount of consumed CH3Cl. They found that in the absence of NOx the final product was HC(O)H in nearly 100% yield and that with NOx in 700 Torr of air HC(O)Cl, HC(O)H and CO were products with yields of (56 ± 10), (32 ± 6) and (12 ± 5)%, respectively. The result of Bilde et al. with NOx contrasts sharply with the large yield of HC(O)Cl and no HC(O)H observed in the reaction of Cl with VA in the presence of NOx presented here. Bilde et al.29 attributed the differences in the yields of HC(O)Cl with and without NOx to the formation of excited Cl–CH2O˙* alkoxy radicals in the exothermic reaction of Cl–CH2OO˙ peroxy radicals with NOx whereas in the NOx-free system only thermalized Cl–CH2O˙ radicals are formed through nearly thermo neutral peroxy–peroxy reactions.
At present it is difficult to explain why we observe mainly formation of HC(O)Cl from the reactions of the Cl–CH2˙ radical formed in the reaction of Cl with VA in the presence of NOx in contrast to the findings of Bilde et al.29 The situation is made even more curious by the fact that in the Cl/AA/NO/air system we observe HC(O)H as the co-product of acetoxyacetaldehyde and not HC(O)Cl. One obvious difference is the source of the Cl–CH2˙ radicals in the studies. Our observations imply that the decomposition channel is not as straightforward as depicted in Fig. 3, i.e. the mechanism is much more complex and may not involve formation of Cl–CH2˙ radicals.
The only structural difference between vinyl acetate and allyl acetate is the –CH2– group in ally acetate between the oxygen atom and the double bond. This difference is structure is apparently critical in determining the reaction products resulting from the terminal –CH2Cl group after consecutive addition of Cl and O2 to form 1,2-chloroperoxy radicals. The carbonyl and C–Cl entities in the intermediate 1,2-chloroperoxy radical reaction intermediates are strongly electron withdrawing. It is possible that there are bonding interactions between the peroxy radicals and the carbonyl and C–Cl entities in the reaction intermediates which complicate the reactions of NO and propably also HO2/RO2 radicals with the 1,2-chloroperoxy radical intermediates and thus the reaction pathways and products. The 1,2-chloroperoxy radical formed in the reaction of Cl with allyl acetate could form a weakly bound six-membered ring with the carbonyl group which would be more favorable than a five-membered ring which could be formed in the reaction of Cl with vinyl acetate. Interaction of the peroxy group with C–Cl and NO could potentially result in the product of chemically activated CH2ClO radicals especially in the case of vinyl acetate where interaction of the peroxy group with the carbonyl group is probably not as important as for allyl acetate. This is presently purely speculation and obviously more detailed laboratory studies as a function of temperature, pressure and O2 and in particular computational studies are needed to shed light on the mechanistic differences observed in the reactions of Cl with vinyl acetate and allyl acetate in the presence and absence of NO.
The other important channel in the reaction of Cl with VA involves an ester rearrangement30 to form CH3C(O)OH with a yield of (27 ± 5)% and the halogenated acyl radical Cl–CH2C˙(O). There are two known fates of acyl radicals either thermal decomposition or addition of O2 to form acylperoxy radicals:31–34
| RC˙(O) + M → R˙ + CO + M |
| RC˙(O) + O2 + M → RC(O)OO˙ + M |
For acyl radicals addition of O2 at atmospheric pressure and room temperature is known to dominate,33,34 however, there is a known monotonic trend towards decomposition, at 298 K and atmospheric pressure of air, with increase in the number of Cl in the CClxF3−xC˙(O) radical.28,29 Although a theoretical study on the atmospheric fate of carbonyl radicals predicts that the main fate of the Cl–CH2C˙(O) radical is addition of O2 and not decomposition the experimental observations in this study of primary CO formation suggests rapid decomposition of the Cl–CH2C˙(O) radical into CO and a Cl–CH2˙ radical rather than addition of O2 (Fig. 3, channel C).
If addition of O2 to Cl–CH2C˙(O) was an important channel the peroxy radicals formed would, due to the high NO2 levels, add NO2 to a significant extent to form the thermally stable PAN type compound ClCH2C(O)OONO2. No evidence could be found in the product spectra for the formation of a PAN type compound.
During this writing a computational study on the mechanism and kinetics of the Cl-initiated oxidation of VA appeared.21 The study found that H-atom abstraction pathways were negligible and that Cl addition dominated followed, in the presence of O2 and NO, by decomposition to give CH3C(O)OC(O)H, CH3C(O)OH and HC(O)Cl as major products as observed in this work. The product channels observed for the reaction of Cl with VA in the presence of NOx are similar to those observed in the analogous OH reaction,20 however, with a higher preference for the channel forming CH3C(O)OC(O)H in the OH reaction.
The results clearly show that in the absence of NOx, only approximately 50% of the reaction is proceeding by the decomposition and rearrangement/decomposition channels of the chloroalkoxy radicals (Fig. 3, channels B and C). As indicated in the Results section the experimental observations support the occurrence of a molecular channel with the formation of chloro-acetic acid anhydride (CH3C(O)OC(O)CH2Cl) through the reaction of the CH3C(O)OCH(O˙)CH2Cl with O2 (Fig. 3, channel D). Based on the carbon balance the yield of chloro-acetic acid anhydride could be potentially as high as 50%. Unfortunately no calibrated reference spectrum is available to verify this.
This result for the reaction of Cl with VA in the absence of NOx contrasts quite markedly with that for the analogous OH reaction under the same conditions. For the reaction of OH with VA in the absence of NOx a molecular channel is not observed. The absence of NOx just results in a stark switch in the preference of the two decomposition channels, i.e. the ester rearrangement channel dominates in the absence of NOx compared to a dominance of the anhydride forming channel in the presence of NOx.20 The substantially lower yields of primary decomposition products for the reaction of Cl with VA in the absence of NOx suggests that chemical activation is important in the atmospheric chemistry of CH3C(O)OCH(O˙)CH2Cl alkoxy radicals.
Cl + allyl acetate
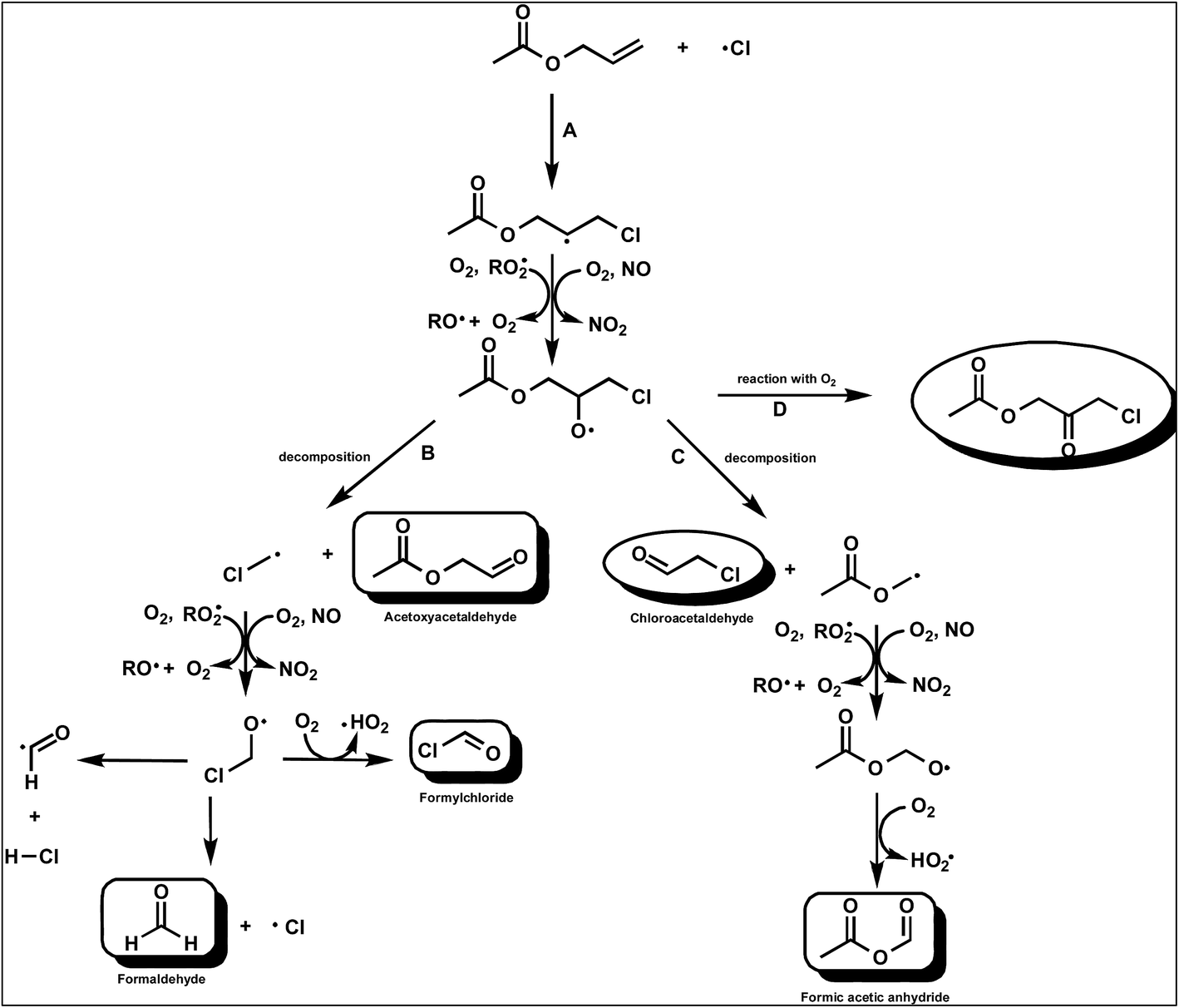 | ||
| Fig. 4 Simplified mechanism for the Cl-atom initiated oxidation of AA (CH2CHCH2OC(O)CH3) via addition of Cl to the terminal carbon of the double bond. | ||
There was no indication for formation of chloroacetaldehyde in the product spectrum showing that channel C is a negligible reaction pathway. As discussed below, from the study on the reaction of Cl with AA in the absence of NOx we are able to put a yield of ≤5% on the formation of CH3C(O)OCH2C(O)CH2Cl from the reaction of the 1,2-chloroalkoxy radicals with O2 (channel D) in the presence of NOx.
In the product study with NOx, acetoxyacetaldehyde, and formaldehyde have been identified as major primary products. The results indicate that the major reaction channel after addition of is cleavage of the C–C bond to form acetoxyacetaldehyde and potentially CH2Cl˙ radicals (Fig. 4, channel B).
As discussed in the Results section although a lower limit of 66% is given in Table 1 for the yield of acetoxyacetaldehyde the observations would suggest that after correction it would be close to unity.
In stark contrast to the reaction of Cl with VA in the presence of NO, HC(O)H is detected as the main co-product originating from the decomposition channel and not HC(O)Cl. As in the VA/Cl/NO/air system, based on the study of Bilde et al.,29 this observation is again in strong contrast to what would be expected from further reactions of the ClCH2˙ radical in the presence of NO. As discussed in the section on the reaction of Cl with NO the decomposition channel as shown in Fig. 4 for the reaction of Cl with AA would appear to be much more complex. Insertion of a –CH2– group between the acetoxy and double bond of VA and AA has resulted in the chlorinated terminal carbon of the compounds producing HC(O)Cl in the case of VA and HC(O)H in the case of AA. The classically accepted mechanism involving C–C cleavage to form CH2Cl˙ radicals can not explain the present product observations in the VA and AA reaction systems.
In addition, to the fast secondary reaction of HCHO in the reaction system, there is obviously also considerable secondary reaction of acetoxyacetaldehyde with Cl. This reaction will proceed mainly through H-atom abstraction from the aldehydic hydrogen:
| CH3C(O)OCH2C(O)H + Cl → CH3C(O)OCH2C˙(O) + HCl |
The resulting acyl radicals are expected, under the experimental conditions, to predominantly add O2 to form peroxy radicals rather than decompose:
| CH3C(O)OCH2C˙(O) + O2 + (M) → CH3C(O)OCH2C(O)OO˙ + (M) |
In the reaction system the peroxy radicals can react with NO or add NO2. Reaction with NO will result mainly in the formation of RC(O)O˙ radicals which lose CO2 and form CH3C(O)OCH2˙ radicals. These radicals undergo further consecutive reactions with O2 and NO to give CH3C(O)OCH2O˙ radicals.
There are two possible reaction pathways for the CH3C(O)OCH2O˙ radical either reaction with O2 to form formic acetic anhydride or an α-ester rearrangement to form acetic acid and HC(O) radicals.
| CH3C(O)OCH2O˙ + O2 → CH3C(O)OC(O)H + HO2 → CH3C(O)OH + HC(O) |
Both acetic acid and formic acetic anhydride have been detected as products in the Cl + AA reaction system in the presence of NOx. We attribute the formation of both of these compounds to secondary reactions of acetoxyacetaldehyde with Cl though the sequence of reactions outlined above. The ratio of the yield of acetic acid to formic acetic anhydride is approximately 4:1 and remains fairly constant throughout the reaction. Picquet-Varrault et al.35 have obtained yields of 75 and 15 for the formation of acetic acid and acetoxyacetaldehyde in the reaction of OH with ethyl acetate which also involves an α-ester rearrangement. The ratio of the two products is quite similar to that obtained in this study for Cl with acetoxyacetaldehyde and we take this as an additional endorsement that secondary reaction of Cl with acetoxyacetaldehyde is the source of acetic acid and formic acetic anhydride in the Cl + AA reaction system. Picquet-Varrault et al.19 in their study on the reaction of OH with AA found acetoxyacetaldehyde as the main product and strongly suspected that secondary reactions of this compound led to the formation of formic acetic anhydride and acetic acid that they observed in their reaction system. This study supports that the reactions of OH and Cl with acetoxyacetaldehyde are similar and result in formation of formic acetic anhydride and acetic acid.
Reaction of the peroxy radicals formed in the reaction of Cl with acetoxyacetaldehyde with NO2 will lead to the formation of a peroxynitrate.
| CH3C(O)OCH2C(O)O2 + NO2 + (M) → CH3C(O)OCH2C(O)O2NO2 + (M) |
This type of peroxynitrate is expected to be reasonably stable at room temperature and atmospheric pressure.36,37 Indeed, bands are present in the product spectrum which can be assigned to a peroxynitrate. Trace A in Fig. 5 shows the spectrum that is obtained at the end of the irradiation of a AA/Cl2/NOx reaction mixture after subtraction of acetoxyacetaldehyde, acetic acid, formic acetic anhydride and nitric acid. The * symbols highlight bands that are characteristic for the formation of a peroxynitrate.36 The band at ∼1744 cm−1 can be assigned to an asymmetric stretching band of NO2 (asym. NO2), that at ∼1330 cm−1 to a symmetrical stretch of NO2 (sym. NO2) and that at ∼795 cm−1 to a NO2 deformation. A band at ∼1045 cm−1 is also observed. Bands in this region are observed in the infrared spectra of many peroxynitrates, however, we have no vibrational group assignment for this band.
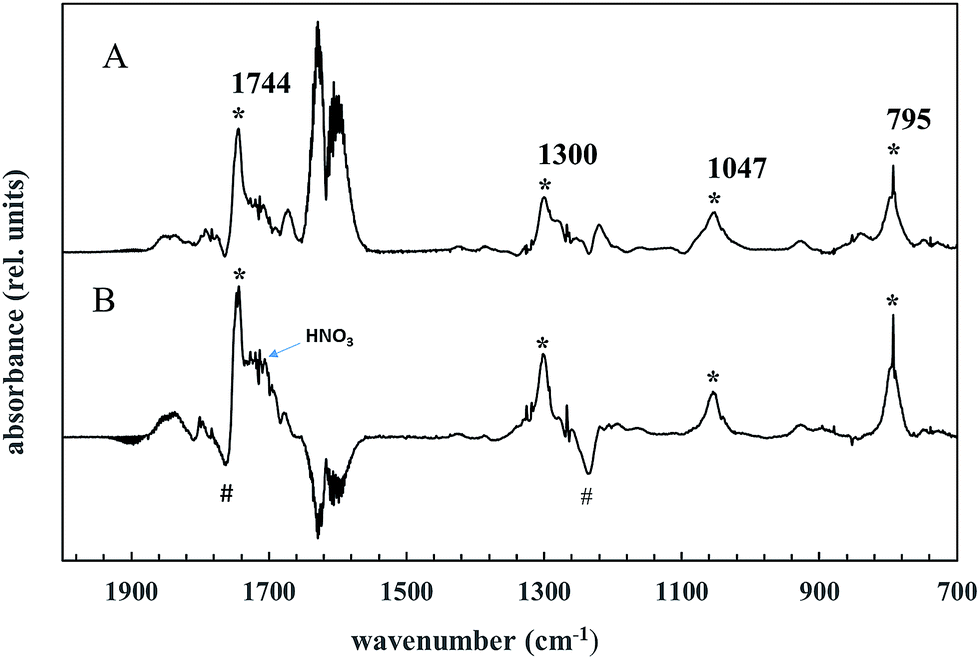 | ||
| Fig. 5 Trace A shows the spectrum obtained at the end of the irradiation a AA/Cl2/NOx reaction mixture after subtraction of acetoxyacetaldehyde, acetic acid, formic acetic anhydride and nitric acid. The * symbols in are attributed to the formation of acetoxyacetyl peroxynitrate. Trace B is the difference spectrum obtained after subtraction of the first spectrum in which the AA was zero from the last spectrum in the experiment. This spectrum demonstrates the secondary decay of the main product acetoxyacetaldehyde (# symbols) and the formation of acetoxyacetyl peroxynitrate (* symbols). | ||
Trace B, Fig. 5 shows a difference spectrum obtained after subtraction of the first spectrum in which the AA concentration was zero from the last spectrum in the experiment. This spectrum shows the continuing secondary decay of the main product acetoxyacetaldehyde (# symbols) and the formation of the bands that we assign to a peroxynitrate. The experimental evidence supports that the peroxynitrate absorptions are arising predominantly from the formation of thermally stable acetoxyacetyl peroxynitrate.
Atmospheric implications
As discussed in several publications the main tropospheric chemical removal of unsaturated compounds is reaction with OH radicals and in the case of VA and AA the tropospheric lifetimes respect to degradation through reaction with OH radicals are approximately 3 and 5 h, respectively,15–20 indicating that these VOCs will be rapidly degraded in the gas phase and near to their emissions sources. The tropospheric lifetimes of the degradation of VA and AA initiated by Cl atoms are 9 and 4 days, respectively.18 However, in marine and industrialised continental regions, where high chlorine concentrations can occur especially at dawn, Cl– mediated degradation of the compounds may be able to compete with the OH– initiated oxidation. Under high NOx conditions both the OH and Cl initiated oxidation of both compounds will give rise to high yields of bond cleavage products including HCHO, HCOCl, CH3C(O)OH, formic acetic anhydride and acetoxyacetaldehyde. Further breakdown of acetoxyacetaldehyde will produce more acetic acid and formic acetic anhydride whereby formation of a potentially thermally stable peroxynitrate is possible which might potentially result in the long range transport of NOx. However, although peroxynitrates of structure ROC(O)OONO2 are thermally stable under atmospheric conditions33 the CH3C(O)O entity in the peroxynitrate formed from acetoxyacetaldehyde, i.e. CH3C(O)OCH2C(O)OONO2, might render this particular susceptible to hydrolysis and thus rapid removal from the atmosphere.Since the anhydrides produced in the atmospheric degradation of VA and AA are not very reactive toward either OH or Cl,38 processes such as washout or heterogeneous hydrolysis on aerosols to form formic and acetic acids are probably the major atmospheric loss processes for these compounds.
Under low NOx conditions only the reaction of OH with VA will lead to the types of bond cleavage products listed above. The reaction of Cl with VA and the reactions of OH and Cl with AA will produce hydroxy acyl or chloro acyl compounds retaining the parent compound carbon skeleton. These compounds will not be very reactive toward OH and their atmospheric degradation, wither in the gas, aqueous or heterogeneous phase will probably result in acid formation, however, this may not necessarily occur close to the emission source.
It is probably safe to say that the both the OH-radical and Cl-atom mediated atmospheric degradation of VA and AA will result primarily in acid formation under both low and high NOx conditions but probably on different time scales.
Acknowledgements
The authors wish to acknowledge the Alexander von Humboldt Foundation, the EU project EUROCHAMP2, DAAD-PROALAR (Germany), MINCyT (Argentina) and CONICET (Argentina) for financial support of this research.References
- T. E. Graedel, D. T. Hawkins and L. D. Claxton, Atmospheric Chemical compounds: Sources, Occurrence and Bioassay, Academic Press, Orlando, FL, 1986 Search PubMed.
- C. Ferrari, Ph.D. thesis, Université Joseph Fourier-Grenoble, 1995.
- J. Rudolph, R. Koppmann and C. Plass-Dülmer, Atmos. Environ., 1996, 30, 1887–1894 CrossRef CAS.
- H. B. Singh, A. N. Thakur, Y. E. Chen and M. Kanakidou, Geophys. Res. Lett., 1996, 23, 1529–1532 CrossRef CAS.
- O. W. Wingenter, D. R. Blake, N. J. Blake, B. C. Sive, F. S. Rowland, E. Atlas and F. Flocke, J. Geophys. Res., 1999, 104, 21–28 CrossRef.
- R. Vogt, P. J. Crutzen and R. A. Sander, Nature, 1996, 383, 327–330 CrossRef CAS PubMed.
- C. W. Spicer, E. G. Chapman, B. J. Finlayson-Pitts, R. A. Plastridge, J. M. Hubbe, J. D. Fast and C. M. Berkowitz, Nature, 1998, 394, 353–356 CrossRef CAS.
- G. Sarwar and P. V. Bhave, J. Clim. Appl. Meteorol., 2007, 46, 1009–1019 CrossRef.
- T. P. Riedel, N. L. Wagner, W. P. Dube, A. N. Middle, C. J. Young, K. F. Öztür, R. Bahreini, T. C. VandenBoer, D. E. Wolfe, E. J. Williams, J. M. Roberts, S. S. Brown and J. A. Thornton, J. Geophys. Res., 2013, 118, 8702–8715 CAS.
- J. A. Thornton, J. P. Kercher, T. P. Riedel, N. L. Wagner, J. Cozic, J. S. Holloway, W. P. Dubé, G. M. Wolfe, P. K. Quinn, A. M. Middlebrook, B. Alexander and S. S. Brown, Nature, 2010, 464, 271–274 CrossRef CAS PubMed.
- G. J. Philips, M. J. Tang, J. Thieser, B. Brickwedde, G. Schuster, B. Bohn, J. Lelieveld and J. N. Crowley, Geophys. Res. Lett., 2012, 39(L10811), 1–5 Search PubMed.
- T. P. Riedel, N. L. Wagner, W. P. Dubé, A. M. Middlebrook, C. J. Young, F. Öztürk, R. Bahreini, T. C. VandenBoer, D. E. Wolfe, E. J. Williams, J. M. Roberts, S. S. Brown and J. A. Thornton, J. Geophys. Res., 2013, 118, 8702–8715 CAS.
- B. J. Finlayson-Pitts, M. J. Ezell and J. N. Pitts, Nature, 1989, 337, 241–244 CrossRef CAS PubMed.
- T. P. Riedel, T. H. Bertram, T. A. Crisp, E. J. Williams, B. M. Lerner, A. Vlasenko, S. M. Li, J. Gilman, J. Gouw, D. M. Bon, N. L. Wagner, S. S. Browm and J. A. Thornton, Environ. Sci. Technol., 2012, 46, 10463–10470 CrossRef CAS PubMed.
- M. B. Blanco, I. Bejan, I. Barnes, P. Wiesen and M. A. Teruel, J. Phys. Chem. A, 2009, 113, 5958–5965 CrossRef CAS PubMed.
- S. Le Calvé, A. Mellouki, G. Le Bras, J. Treacy, J. Wenger and H. Sidebottom, J. Atmos. Chem., 2000, 37, 161–172 CrossRef.
- B. Picquet-Varrault, M. Scarfogliero and J. F. Doussin, Environ. Sci. Technol., 2010, 44, 4615–4621 CrossRef CAS PubMed.
- M. B. Blanco, I. Bejan, I. Barnes, P. Wiesen and M. A. Teruel, Environ. Sci. Pollut. Res., 2009, 16, 641–648 CrossRef CAS PubMed.
- B. Picquet-Varrault, J. F. Doussin, R. Durand-Jolibois, O. Pirali and P. Carlier, Environ. Sci. Technol., 2002, 36, 4081–4086 CrossRef CAS.
- M. B. Blanco, I. Bejan, I. Barnes, P. Wiesen and M. A. Teruel, Environ. Sci. Technol., 2012, 46, 8817–8825 CrossRef CAS PubMed.
- J. Li, H. Cao, D. Han, M. Li, X. Li, M. He and S. Ma, Atmos. Environ., 2014, 94, 63–73 CrossRef CAS PubMed.
- I. Barnes, K. H. Becker and N. Mihalopoulos, J. Atmos. Chem., 1994, 18, 267–289 CrossRef CAS.
- J. F. Doussin, D. Ritz, R. D. Jolibois, A. Monod and P. Carlier, Analysis, 1997, 35, 236–242 Search PubMed.
- E. C. Tuazon, H. Mac Leod, R. Atkinson and W. P. L. Carter, Environ. Sci. Technol., 1986, 20, 383–387 CrossRef CAS PubMed.
- W. B. DeMore, S. P. Sander, D. M. Golden, R. F. Hampson, M. J. Kurylo, C. J. Howard, A. R. Ravishankara, C. E. Kolb and M. J. Molina, Evaluation Number 12, JPL Publ., 1997, 97–4.
- J. J. Orlando, G. S. Tyndall and T. J. Wallington, Chem. Rev., 2003, 103, 4657–4689 CrossRef CAS PubMed.
- C. M. Butt, C. J. Young, S. A. Mabury, M. D. Hurley and T. J. Wallington, J. Phys. Chem. A, 2009, 113, 3155–3161 CrossRef CAS PubMed.
- M. Walavalkar, A. Sharma, H. D. Alwe, K. K. Pushpa, S. Dhanya, P. N. Naik and P. N. Bajaj, Atmos. Environ., 2013, 67, 93–100 CrossRef CAS PubMed.
- M. Bilde, J. J. Orlando, G. Tyndall, T. J. Wallington, M. D. Hurley and E. W. Kaiser, J. Phys. Chem. A, 1999, 103, 3963–3965 CrossRef CAS.
- E. C. Tuazon, S. M. Aschmann, R. Atkinson and W. P. L. Carter, J. Phys. Chem. A, 1998, 102, 2316–2321 CrossRef CAS.
- I. Barnes, K. H. Becker, F. Kirchner, F. Zabel, H. Richer and J. Sodeau, STEP-HALOCSIDE/AFEAS Workshop, March 23-25, Dublin, 1993 Search PubMed.
- E. C. Tuazon and R. Atkinson, J. Atmos. Chem., 1993, 16, 301–312 CrossRef CAS.
- S. Jagiella, H. G. Libuda and F. Zabel, Phys. Chem. Chem. Phys., 2000, 2, 1175–1181 RSC.
- R. I. Méreau, M. T. Rayez, J. C. Rayez, F. Caralp and R. Lesclaux, Phys. Chem. Chem. Phys., 2001, 3, 4712–4717 RSC.
- B. Picquet-Varrault, J. F. Doussin, R. Durand-Jolibois and P. Carlier, Phys. Chem. Chem. Phys., 2001, 3, 2595–2606 RSC.
- F. Kirchner, Kinetische Untersuchungen an Peroxynitraten und Peroxy-Radikalen, PhD thesis, University of Wuppertal, Germany, 1994.
- F. Kirchner, L. P. Thüner, I. Barnes, K. H. Becker, B. Donner and F. Zabel, Environ. Sci. Technol., 1997, 31, 1801–1804 CrossRef CAS.
- J. G. Calvert, A. Mellouki, J. J. Orlando, M. J. Pilling and T. J. Wallington, Mechanisms of atmospheric oxidation of the oxygenates, Oxford University Press, New York, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra04975j |
| This journal is © The Royal Society of Chemistry 2015 |