
Influence of earth-abundant bimetallic (Fe–Ni) nanoparticle-embedded CNFs as a low-cost counter electrode material for dye-sensitized solar cells
Abstract
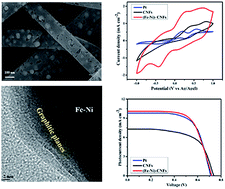
Earth-abundant bimetallic (Fe–Ni) nanoparticle-embedded carbon nanofibers (CNFs) have been prepared by electrospinning technique and used as counter electrode (CE) material for dye-sensitized solar cells (DSSC). The structural and morphological properties were explored by X-ray diffraction (XRD), Raman spectroscopy, field-emission scanning microscope (FE-SEM) and transmission electron microscope (TEM) studies. The results of cyclic voltammetry (CV), electrochemical impedance and Tafel polarization studies revealed that (Fe–Ni)-CNFs demonstrated superior electrocatalytic activity, electrochemical stability, low charge transfer resistance (Rct) and high exchange current density for the reduction of triiodide (I3−). Furthermore, it was observed that DSSC fabricated using (Fe–Ni)-CNFs as counter electrode had achieved power conversion efficiency (PCE) that is almost comparable to the same fabricated using std. Pt. This is due to the prepared CNFs' large surface area and randomly oriented, interconnected porous morphology with graphitized structure, which enhanced the contact with a large quantity of ionic liquid electrolyte, leading to a faster redox kinetics of I3−/I−. This study revealed that (Fe–Ni)-CNFs could be used as a low-cost and efficient counter electrode material for DSSCs.
 Please wait while we load your content...
Please wait while we load your content...

