
Theoretical investigation of dihydroacridine and diphenylsulphone derivatives as thermally activated delayed fluorescence emitters for organic light-emitting diodes†
Xiaguang Zhang,
Wei Shen,
Dongmei Zhang,
Yongzhen Zheng,
Rongxing He and
Ming Li*
School of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, P. R. China. E-mail: liming@swu.edu.cn; Tel: +86-023-68253023
First published on 1st May 2015
Abstract
A series of donor–acceptor–donor type compounds containing 9,9-dimethyl-9,10-dihydroacridine and diphenylsulphone as thermally activated delayed fluorescence emitters are designed and investigated, and their broad application prospects in organic light emitting diodes are predicted by density functional theory (DFT). The results show that the orbital interaction of the atom between the acceptor and the donor is an important factor to influence the singlet–triplet energy difference. Effective intermolecular singlet–singlet, singlet–triplet and triplet–triplet energy transfers from hosts to emitters, in which donors and acceptors are linked by C–N bonds, would occur. The para- and meta-linked compounds exhibit blue emission and the ortho-linked compounds show green emission in these emitters.
1. Introduction
In organic light-emitting diodes (OLEDs), the holes and electrons recombine to produce singlet and triplet excitons and the ratio of spin statistics is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3,1,2 so only a 25% internal quantum efficiency would be harvested in fluorescent OLEDs.3 Recently, a thermally activated delay fluorescence (TADF) mechanism has been applied to OLEDs.4,5 In TADF-OLEDs, the organic emitters, which have a small singlet and triplet difference (ΔEST), can provide a reverse intersystem crossing (RISC) and effectively transform both singlet and triplet excitons into light, reaching 100% maximum internal quantum efficiency.6–8 Compared with phosphorescent OLEDs, the TADF-OLEDs have a strong advantage in reducing the high costs of valuable transition metals, balancing the charge carrier transport and solving the instability of blue transition metal phosphors.9 Recently, the study of TADF-OLEDs material has attracted considerable attention. However, developing a TADF emitter is a challenging task. On the one hand, the compound requires a sufficiently small ΔEST to ensure that triplet excitons can transfer from triplet state to singlet state. On the other hand, it needs appropriate energy level matching of singlet and triplet energy between host and TADF emitter. An understanding of the structure–property relationships in TADF emitters is essential for solving the abovementioned problem.
:3,1,2 so only a 25% internal quantum efficiency would be harvested in fluorescent OLEDs.3 Recently, a thermally activated delay fluorescence (TADF) mechanism has been applied to OLEDs.4,5 In TADF-OLEDs, the organic emitters, which have a small singlet and triplet difference (ΔEST), can provide a reverse intersystem crossing (RISC) and effectively transform both singlet and triplet excitons into light, reaching 100% maximum internal quantum efficiency.6–8 Compared with phosphorescent OLEDs, the TADF-OLEDs have a strong advantage in reducing the high costs of valuable transition metals, balancing the charge carrier transport and solving the instability of blue transition metal phosphors.9 Recently, the study of TADF-OLEDs material has attracted considerable attention. However, developing a TADF emitter is a challenging task. On the one hand, the compound requires a sufficiently small ΔEST to ensure that triplet excitons can transfer from triplet state to singlet state. On the other hand, it needs appropriate energy level matching of singlet and triplet energy between host and TADF emitter. An understanding of the structure–property relationships in TADF emitters is essential for solving the abovementioned problem.
For TADF-OLEDs emitter, the type of donor–acceptor (D–A) type or donor–acceptor–donor (D–A–D) is a trends in current research.10,11 Generally, there is a small spatial overlap between the HOMO and the LUMO for donor and acceptor structures, which can contribute to induce intramolecular charge transfer that leads to a small exchange integral between donor and acceptor and causes a small singlet–triplet splitting, namely, a small ΔEST. Based on this concept, Adachi's group has chosen various donors and acceptors to design TADF emitters to be applied in OLED, and consequently, a high external electroluminescence quantum efficiency (EQE) is achieved.12–14 Among the synthesized TADF materials, diphenylsulphone and acridine derivatives are always used for donor and acceptor, and they showed excellent performances. For example, bis[4-(3,6-dimethoxycarbazole)phenyl]sulfone has a small ΔEST (0.21 eV). As an emitter, the OLED device shows an excellent maximum EQE of 14.5% and reduces efficiency roll-off.15 The highly efficient TADF performance of spiro-acridine has been demonstrated, and it exhibits a high internal photoluminescence efficiency value (67.3%).16 A device with 9,10-dihydroacridine and a diphenylsulphone derivative as emitter can exhibit a high EQE (19.5%), which makes it comparable with the best phosphorescent OLEDs available.12 In addition, Adachi's group demonstrated that a small ΔEST and a large fluorescence rate (kF) can be achieved by increasing the distance between donor and acceptor in intramolecular-charge-transfer molecules.17 At the same time, many studies show that increasing the steric hindrance between donor and acceptor can also lead to the spatial separation of the frontier orbitals, and the higher photoluminescence quantum yields appeared in the D–A–D-type molecules rather than the D–A-type molecules.18,19
Herein, we have performed density functional theory (DFT) calculations to investigate a series of TADF-emitters based on a D–A–D electronic structure, comprising diphenylsulphone electron acceptor core units substituted with electron donor units of 9,9-dimethyl-9,10-dihydroacridine (Fig. 1). In addition, to verify the molecular properties of the TADF emitters described above, m-bis(N-carbazolyl)benzene (mCP), 4,4′-bis(carbazole-9-yl)biphenyl (CBP) and bis(2-(diphenylphosphino)phenyl)ether oxide (DPEPO) are selected as reference host materials. In this work, we theoretically predict the application prospects of the studied compounds in TADF-OLEDs based on the ΔEST and intermolecular energy transfer between host and emitter. Our goal is to investigate the TADF mechanism of energy transfer in the host–guest system and to analyze the influence of the different linkage styles of carbon–nitrogen (para-, meta- and ortho-) and carbon–carbon (para-, meta- and ortho-) between donors and acceptors on molecular performance.
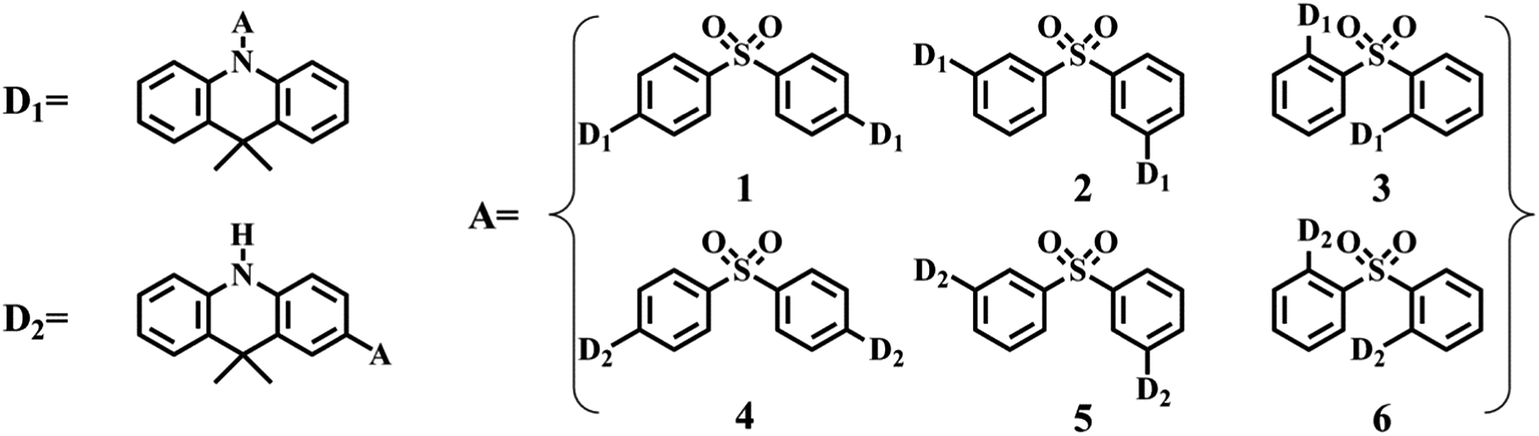 | ||
| Fig. 1 Molecular structures of compounds 1–6. | ||
2. Computation details
In this work, in order to select a reliable method to study the ground and excited states of the investigated system, different density functional theory (DFT) methods were employed with 6-31G(d) and 6-311+G(d) basis sets (the results are listed in the ESI†). Frequency calculations showed that the optimized structures have the minimum energy states on the potential energy surfaces. In this study, the maximum deviation (MAX), the mean absolute deviation (MAD), the root mean-squared deviation (RMSD), and the relative error (Er) have been used as a guide for comparison between the experimental and the calculated values.20 The parameters are defined as
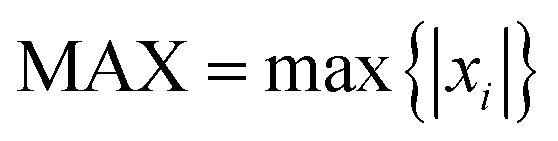 | (1) |
 | (2) |
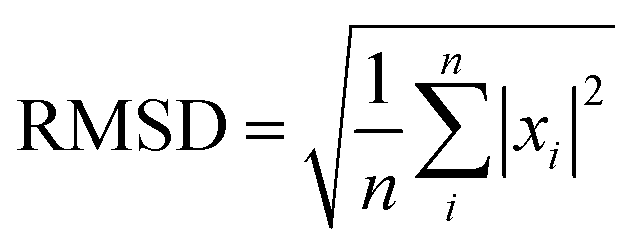 | (3) |
 | (4) |
The impact on the geometries of the ground (S0) and the triplet (T1) states with different functionals and basis sets (Table S1†) were discussed. The geometry of the T1 state is optimized via the spin-unrestricted DFT level, and the triplet energy (ET) is also corrected for zero-point energies. With a 6-31G(d) basis set, the HOMO and the ET produced the smallest MAX, MAD and RMSD (0.01, 0.00 and 0.007, respectively) with a B3P86 functional. With a 6-31+G(d) basis set, the HOMO produced the smallest Er with the M06 method. Accordingly, the calculated results at the B3P86/6-31G(d) level21 are consistent with the experimental data (Table S2†). Generally, it is difficult to describe the virtual orbitals theoretically than occupied orbitals. If the main transition of the first excitation state originates from a HOMO to LUMO promotion, the LUMO energy can be obtained from the HOMO energy plus the first vertical excitation energy. Vertical absorption and emission energies can be obtained from a time-dependent density functional theory (TD-DFT) calculation based on the optimized S0 and the first singlet excited (S1) state geometries, respectively. By combining the HF exchange percentage (HF%)22 and the impact of the solvent effect, many functionals, which possess different HF% values in toluene (by using the polarized continuum model (PCM)23) and in the gas phase, were performed (Table S1 and S3†). One can find that the results with TD-B3LYP/6-31G(d)//B3P86/6-31G(d)24 in toluene can obtain more accurate excitation energies and LUMO energies when compared with the experimental data (the MAD and RMSD are 0.047 and 0.047, respectively). From the results in Table S4,† which are calculated using TD-B3LYP/6-31G(d) in toluene, one can obtain a more accurate emission wavelength (Er = 2.14%). All the calculations are performed using the Gaussian09 program.25
3. Results and discussion
3.1. Geometry structures of the optimized ground and singlet excited states
The structure parameter of a compound is a key factor that contributes to the understanding of the radiative transition of the molecule; the selected bond lengths and the dihedral angles of compounds in ground (S0) and singlet excited (S1) states are collected in Table 1, S5 and Fig. S1.† For the C–N linkage series, the dihedral angle between the donor (9,9-dimethyl-9,10-dihydroacridine) and the phenyl plane of the acceptor (diphenylsulphone) is over 75°, which shows that the steric effects in the ground states are far away from 0°. A large dihedral angle could limit the coupling between the donor and the acceptor in the S0 state. The natural bond orbital analysis shows that the nitrogen atoms adopt an sp2 hybridized orbital, while the bond between the nitrogen atom (donor) and the carbon atom (acceptor) is a weak p–π conjugation, leading the bond length of nitrogen–carbon between the nitrogen–carbon single bond length (1.54 Å) and the nitrogen–carbon conjugation bond length (1.37 Å), which indicates a small bond energy. Due to the high binding energy between donor and acceptor, a linkage bond, which has a length between the carbon–carbon single bond length (1.54 Å) and the carbon–carbon double bond length (1.34 Å), in the C–C linkage series is found to be comparable with the C–N linkage series. In addition, the linkage position (ortho-, meta-, para-) has a great influence on the structure parameter of the C–C linkage series (dihedral angle para- ≈ meta- > ortho-). In the excited state, the steric effect also limits the flattening distortion of compounds in the C–N linkage series, whereas the C–C linkage series has an evident planarization tendency. Higher Franck–Condon factor will be formed accompanied with the structural deformation of the excited and ground states, which leads to a highly effective nonradiative relaxation channel.26,27 Results showed that the radiative transition properties of the C–N linkage series are more effective than those of the C–C linkage series.| Compound | Bond length/Å | Dihedral angle/° | |||
|---|---|---|---|---|---|
| 1 | S0 | 1.4265 | 1.4265 | −76.4372 | −77.7945 |
| 2 | S0 | 1.4260 | 1.4261 | 77.6548 | 78.5084 |
| 3 | S0 | 1.4265 | 1.4265 | −77.1852 | −77.1719 |
| 4 | S0 | 1.4748 | 1.4751 | −35.1964 | 35.0320 |
| 5 | S0 | 1.4762 | 1.4763 | −35.5985 | 34.9405 |
| 6 | S0 | 1.4859 | 1.4857 | −61.3863 | −55.1947 |
In the ground state, the large torsion angle between the donor and the acceptor plays an important role in separating the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). The HOMO and the LUMO of these compounds are depicted in Fig. 2. Due to the large dihedral angle between the donor and the acceptor, the molecules in the C–N linkage series show a small spatial overlap between the HOMO and the LUMO. The HOMO of the C–C linkage series tends to be delocalized over the conjugated area, and thus the HOMO and the LUMO exhibit a large orbital overlap. In addition, the density of states and projected density of states are calculated to test the orbital overlap degree; related results are shown in Fig. S2.† For the C–N linkage series, the densities of the HOMO are localized on the donor and occupation values for the electronic transitions model exceed 95% (para-, meta- and ortho- are 97.3%, 97.0% and 96.3%, respectively), whereas the densities of the LUMO are localized on the acceptor and occupation values of para-, meta- and ortho- are 96.8%, 97.8%, 97.2%, respectively. For the C–C linkage series, the donor occupation values of the HOMO for para-, meta- and ortho- are 89.9%, 92.1% and 95.9%, respectively, and the corresponding occupation values of the LUMO, localized on the acceptor, are 75.3%, 92.0%, 94.2%, respectively. The results show that the dihedral angle increases as the overlap degree of the HOMO and the LUMO decreases, for the C–C linkage series (overlap degree para- > meta- > ortho-).
 | ||
| Fig. 2 Frontier molecular orbitals for compounds 1–6 in the ground state. | ||
Based on the abovementioned analysis, C–N linkage series have larger torsion angles between donor and acceptor than C–C linkage series and show a smaller spatial overlap between the HOMO and the LUMO.
3.2. Singlet and triplet excitation energy
In order to examine the electronic transition nature of compounds, TD-DFT calculations are implemented. The results are collected in Table S6;† as shown, the HOMO → LUMO character has predominated in the transition from S0 → S1. In general, the natural transition orbital is equal to the HOMO and LUMO when the probability is over 90%, and the natural transition orbitals for the S0 → S1 and S0 → T1 transitions are displayed in Fig. S3.† For the C–N linkage series, the hole is located on the donor and the particle is located on the acceptor. For the C–C linkage series, the holes and the particles in the compounds have relatively large orbital overlaps, and the results are similar to the HOMO–LUMO analysis. The transition state of the S0 → S1 transition in the C–N linkage series is an evident intramolecular charge transfer state, so compounds 1–3 have a small transition energy for the S0 → S1 transition. In general, it is necessary for an efficient thermally activated delayed fluorescence (TADF) emitter that there is a small energy difference between the first excited singlet and triplet states (that is ΔEST). The ΔEST values of the compounds are collected in Table 2. The values of ΔEST in the C–N linkage series are considerably less than those in the C–C linkage series; the small orbital overlap between hole and particle in the C–N linkage series leads to the low orbital overlap integral between ϕ and ϕ*, that is, a weak exchange integral J (ϕ, ϕ*). As shown in eqn (5),28–30 the degree of spatial overlap will strongly alter the value of ΔEST. The up-conversion process of the T1 → S1 transition in the C–N linkage series is thermodynamically favorable, because the small ΔEST enables a faster reverse intersystem crossing (RISC) rate for the T1 →S1 transition than for the non-radiative decay of the T1 → S0 transition. According to the energy gap law, the rate of internal conversion exceeds the intersystem crossing rate or the RISC rate, so the RISC occurs between the S1 and T1 states. As the ΔEST of the C–C linkage series is large, the up-conversion from T1 to S1 is difficult. In addition, the compounds also possess a high ΔEST in the higher-order excited states, which means that for the compounds in the C–C linkage series TADF would not occur.
 | (5) |
| Compound | HOMO | LUMOa | ET | E(S1) | E(T1) | ΔEST |
|---|---|---|---|---|---|---|
| a LUMO = E(S1) − HOMO.b Experimental date (ref. 12).c Donor is 9,9-dimethyl-9,10-dihydroacridine.d Acceptor is diphenylsulphone. | ||||||
| 1 | −5.92 | −2.97/2.92b | 2.92 | 2.95 | 2.94 | 0.01 |
| 2 | −5.88 | −2.89 | 2.98 | 2.99 | 2.97 | 0.02 |
| 3 | −5.73 | −2.86 | 2.69 | 2.87 | 2.85 | 0.02 |
| 4 | −5.86 | −2.59 | 2.91 | 3.27 | 2.78 | 0.49 |
| 5 | −5.83 | −2.39 | 2.67 | 3.44 | 2.92 | 0.52 |
| 6 | −5.77 | −2.24 | 2.84 | 3.53 | 3.15 | 0.38 |
| mCP | −6.08 | −2.06 | 3.19/2.90b | 4.02 | 3.21 | 0.81 |
| DPEPO | −6.65 | −1.94 | 3.25/3.30b | 4.71 | 3.59 | 1.12 |
| CBP | −5.98 | −2.38 | 2.73/2.64b | 3.60 | 2.99 | 0.61 |
| Donorc | −5.74 | −1.36 | 3.29 | 4.38 | 3.43 | 0.95 |
| Acceptord | −7.78 | −2.65 | 3.92 | 5.13 | 3.64 | 1.49 |
As is known to all, the small ΔEST, which resulted from the large dihedral angle between the donor and the acceptor of the compounds, may lead to the occurrence of an efficient TADF emission. Why does compound 6, which possesses a large dihedral angle (∼60°) between donor and acceptor, have a large ΔEST? This can be explained by the stronger electron donating ability of the molecules with the C–N linkage (the natural population analysis charges of the donors of compounds 1–3 and 6 are 0.39, 0.38, 0.37 and 0, respectively). In addition, we also scanned the torsion angles between the donors and the acceptors of compounds 1–3 and 6 to explore their influence on the value of ΔEST.31 The results are collected and shown in Table S7.† In compounds 1–3, when the dihedral angles approximate to 0°, the HOMO is localized on the full molecular structure and the LUMO is mainly localized on the π*-orbitals of the acceptor. In addition, the excited singlet states are largely constructed from HOMO → LUMO transitions. The dihedral angle has a great influence on the energy of the S0 → S1 transition, and the large spatial overlap leads to a large ΔEST. At the same time, the small dihedral angle leads to a strong p–π conjugation of nitrogen–carbon between the donor and the acceptor, which helps to increase the intramolecular interaction. Consequently, for compounds 1–3, no TADF emission occurs when the dihedral angles approximate to 0°. For compound 6, when the dihedral angle is 0°, the HOMO is localized on the full molecular structure and the LUMO is mainly localized on the acceptor. The ΔEST is 0.46 eV, and this value is similar to the ΔEST of compound 6 when the dihedral angle is 60°. When the dihedral angle is 90°, the large spatial overlap between the HOMO and the LUMO is also realized, leading to a large ΔEST, and thus, no TADF emission occurs. The carbon–carbon bond has a large π–π conjugation, no matter how large the dihedral angle is between the donor and the acceptor in compound 6. Namely, the significant spatial overlap leads to a large ΔEST.
In summary, the C–C linkage series compounds have a large ΔEST, which makes them unsuitable as TADF emitters. In addition, the orbital interaction of the atom between the acceptor and the donor is an essential factor, which has an impact on the value of ΔEST.
3.3. Energy and charge transfer between emitter and host
In order to avoid concentration quenching and ensure highly efficient electroluminescence, the TADF emitters are usually dispersed in a suitable host material, which has a higher triplet energy (ET) than that of the emitter to avoid reversing the energy transfer from the host to the emitter.32 In this work, mCP, CBP and DPEPO are selected as host materials. As the compounds in the C–C linkage series are unsuitable as TADF emitters, we only discuss the compounds in the C–N linkage series. Table 2 shows that the ET of the host molecule, CBP (2.73 eV), is smaller than that of the emitter compounds, 1 (2.92 eV) and 2 (2.98 eV), so compounds 1 and 2 dispersed in CBP are unsuitable. Moreover, the results also verify the experimental conclusion that the PLQY of compound 1 increases when the host is changed from CBP to mCP to DPEPO.12 In general, singlet and triplet excitons are generated by electron–hole recombination in the host. Singlet excitons from the host transfer into the singlet state of the emitter by the Förster energy transfer process.33 Triplet excitons from the host are transferred into the singlet state or the triplet state of the emitter by the Dexter energy transfer process.34 The transition of T1 → S0 in the host molecules, CBP, mCP and DPEPO, is spin forbidden, and the calculated transition dipole moment (Table S8†) of the host shows that the transition S1 → S0 of DPEPO is allowed, which also verifies the experimental conclusion. The transition energies of S1 and T1 in the host and the emitter (Table 2) show that the S1 → S1 intermolecular Förster energy transfer from DPEPO to emitters 1, 2 and 3, which is thermodynamics favorable and engender prompt fluorescence emission. As can be seen from the transition energy, the singlet excitation energy E(S1) and the triplet excitation energy E(T1) of emitters 1, 2 and 3 are smaller than the E(T1) of the host molecule, DPEPO, which demonstrates that the T1 → S1 Förster energy transfer and the T1 → T1 Dexter energy transfer from DPEPO to the emitters is also thermodynamically favorable. Moreover, emitters 1 and 3 dispersed in the host molecules, CBP and mCP, respectively. This could engender S1 → S1 intermolecular Förster energy transfer and T1 → T1 Dexter energy transfer, when viewed from a thermodynamics point of view. In addition, the substantial overlap of the host emission and the emitter absorption could effectively demonstrate the occurrence of Förster energy transfer, which is a long-range energy transfer. Fig. 3 shows that the absorption wavelength of the emitters (1, 2 and 3 are 420.17 nm, 414.18 nm and 431.50 nm, respectively) and the emission wavelength of DPEPO (403.72 nm) show substantial spectral overlap between host emission and emitter absorption.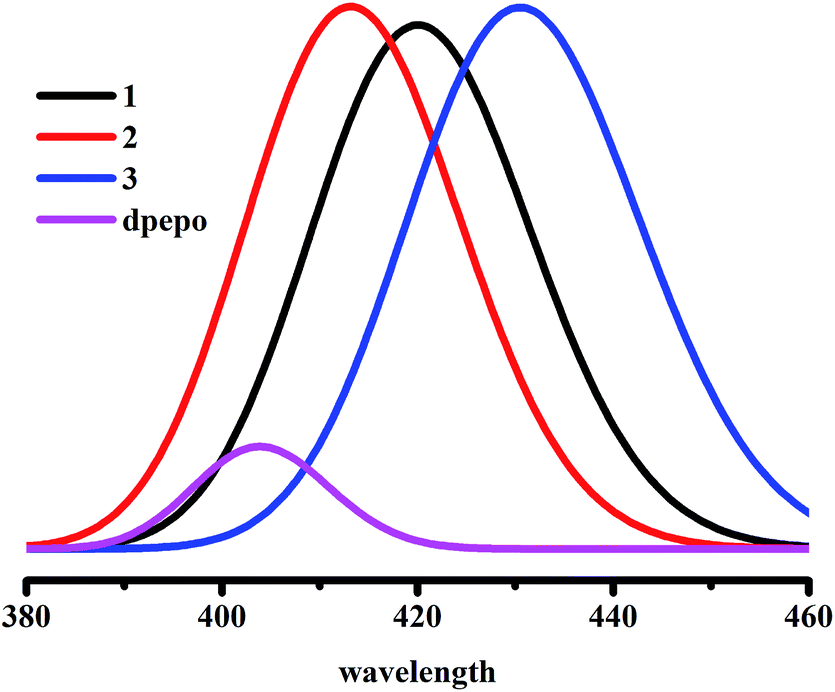 | ||
| Fig. 3 Emitters' (1, 2 and 3) absorption spectra and host's (DPEPO) emission spectra. | ||
In addition, balanced charge transport and fast charge transport rates are important for the TADF emitter performance. The Marcus–Hush equation shows that a low reorganization energy (λ) and a high transfer integral (V) could lead to a high charge mobility rate35 and λ is a main influential factor. Table S9† shows that the monotonous increase of λh in the C–N linkage series is 1 (para-) < 2 (meta-) < 3 (ortho-), namely, that the hole transfer rate is 3 (ortho-) < 2 (meta-) < 1 (para-), while the monotonous increase of λe and Ve in the C–N linkage series in the order 1 (para-) < 2 (meta-) < 3 (ortho-) and 3 (ortho-) < 2 (meta-) < 1 (para-), respectively, means that the electron transfer rate is 3 (ortho-) < 2 (meta-) < 1 (para-). The results show that compound 1 (para-) has the fastest charge transfer rate of the C–N linkage series compounds. The values of λh/λe increase as follows: 3 (ortho-) < 2 (meta-) < 1 (para-), demonstrating that compound 1 (para-) exhibits a good balanced charge (hole and electron) transport capacity.
From the abovementioned discussion, DPEPO as a host molecule suited all emitters and CBP also conforms to the requirements of emitter 3. In addition, effective energy transfer could occur between host and emitter. Moreover, the C–N linkage series has a good balance of charge transport performance and fast charge transport rate; in addition, compound 1 (para-) exhibits the best charge transport performance.
3.4. Potential device performances used in OLED
A low energy barrier for charge injection from the neighboring layers is the basic requirement for the design of an OLED, so higher HOMO and lower LUMO energy levels than those of the host are desired for the emitter to ensure efficient direct charge trapping on the emitting layer. Compared with the host molecule, DPEPO, the energies of the frontier molecular orbitals (Table 2) of the C–N linkage series meet the direct charge trapping requirement.36 The energy of the HOMO level increases monotonously in these compounds (3 (ortho-) < 2 (meta-) < 1 (para-)). Hence, hole transfer from host DPEPO to emitter 3 would be more facile than with other emitters. For electron injection, the changes of energy of the LUMO level are not evident and they increase monotonously in these compounds (1 (para-) < 2 (meta-) < 3 (ortho-)), and 2 (−2.89 eV) and 3 (−2.86 eV) have similar LUMO energy levels. The results show that all the compounds have similar electron-injection barriers. Furthermore, ionization potentials (IP) and electron affinities (EA) are consistent with the abovementioned result (Table S9†). Thus, compound 3 (ortho-) has an appropriate charge injection performance.The different emitters (1, 2 and 3) also show a good potential performance due to the small ΔEST which is correlate to a nice device performance. Table 3 shows that the maximum emission wavelengths of compounds 1, 2 and 3 are 471.08 nm, 475.19 nm and 617.28 nm, respectively. Compounds 1 and 2 exhibit a blue emission and 3 exhibits a green emission.
| Compound | Wavelength | E |
|---|---|---|
| 1 | 471.08 | 2.63 |
| 2 | 475.19 | 2.61 |
| 3 | 617.28 | 2.11 |
From the combination of the charge injection barrier and device turn-on voltage, all compounds show good performances when chosen as blue or green TADF-OLED emitters. Moreover, among the C–N linkage series in our work, compound 1 appears to be the most promising blue emission molecule and 3 is a good green emission molecule.
4. Conclusion
In this work, a comprehensive theoretical investigation has been performed to study the D–A–D electronic structure of the C–N linkage series and the C–C linkage series. According to the geometry structure between ground state and singlet excited state, the values of ΔEST and the orbital overlaps of donors and acceptors in the S0 → S1 transition in the two series of compounds show that the C–C linkage series of compounds could not suit to be TADF emitter. The results of the scan torsion angles between donors and acceptors show that the most influential factor on the value of ΔEST is the orbital interaction of the atom between the acceptor and the donor. From the triplet energies and the transition energies of the emitters and the host analysis, we can see that DPEPO is the most suitable host molecule for all series emitter of C–N linkage compounds, and effective energy transfer of S1 → S1, T1 → S1 and T1 → T1 from DPEPO to emitters would occur. Moreover, from the HOMO–LUMO energy, it could be inferred that direct charge trapping would occur on the emitter layer. Furthermore, a good performance and a high RISC for the C–N linkage series compounds can be expected due to the low ΔEST. Of the C–N linkage series compounds, 1 (para-) and 2 (meta-) exhibit blue emission and 3 (ortho-) exhibits green emission. We believe that our study could provide instructive information for experimental and theoretical investigation in TADF-OLED.References
- C. W. Tang and S. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS PubMed.
- M. Pope, H. Kallmann and P. Magnante, J. Chem. Phys., 1963, 38, 2042–2043 CrossRef CAS PubMed.
- M. Baldo, D. O'brien, Y. You, A. Shoustikov, S. Sibley, M. Thompson and S. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS PubMed.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., 2014, 126, 6520–6524 CrossRef PubMed.
- R. Ishimatsu, S. Matsunami, T. Kasahara, J. Mizuno, T. Edura, C. Adachi, K. Nakano and T. Imato, Angew. Chem., Int. Ed., 2014, 53, 6993–6996 CrossRef CAS PubMed.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS PubMed.
- M. Uejima, T. Sato, M. Detani, A. Wakamiya, F. Suzuki, H. Suzuki, T. Fukushima, K. Tanaka, Y. Murata and C. Adachi, Chem. Phys. Lett., 2014, 602, 80–83 CrossRef CAS PubMed.
- T. Komino, H. Tanaka and C. Adachi, Chem. Mater., 2014, 26, 3665–3671 CrossRef CAS.
- S. Wu, M. Aonuma, Q. Zhang, S. Huang, T. Nakagawa, K. Kuwabara and C. Adachi, J. Mater. Chem. C, 2013, 2, 421–424 RSC.
- G. Méhes, H. Nomura, Q. Zhang, T. Nakagawa and C. Adachi, Angew. Chem., Int. Ed., 2012, 51, 11311–11315 CrossRef PubMed.
- Q. Zhang, H. Kuwabara, W. J. Potscavage, S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070–18081 CrossRef CAS PubMed.
- J. Lee, K. Shizu, H. Tanaka, H. Nomura, T. Yasuda and C. Adachi, J. Mater. Chem. C, 2013, 1, 4599–4604 RSC.
- W. Li, Y. Pan, R. Xiao, Q. Peng, S. Zhang, D. Ma, F. Li, F. Shen, Y. Wang and B. Yang, Adv. Funct. Mater., 2014, 24, 1609–1614 CrossRef CAS PubMed.
- M. Moral, L. Muccioli, W.-J. Son, Y. Olivier and J. C. Sancho-García, J. Chem. Theory Comput., 2014, 11, 168–177 CrossRef.
- J. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7406 CrossRef.
- S. Huang, Q. Zhang, Y. Shiota, T. Nakagawa, K. Kuwabara, K. Yoshizawa and C. Adachi, J. Chem. Theory Comput., 2013, 9, 3872–3877 CrossRef CAS.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- N. Turro, Modern molecular photochemistry, Menlo Park, CA, 1978, pp. 317–319 Search PubMed.
- C. L. Linfoot, M. J. Leitl, P. Richardson, A. F. Rausch, O. Chepelin, F. J. White, H. Yersin and N. Robertson, J. Inorg. Chem., 2014, 53, 10854–10861 CrossRef CAS PubMed.
- N. J. Turro, Modern molecular photochemistry, University Science Books, 1991 Search PubMed.
- T. Ziegler, A. Rauk and E. J. Baerends, Theor. Chim. Acta, 1977, 43, 261–271 CrossRef CAS.
- Y. Pan, W. Li, S. Zhang, L. Yao, C. Gu, H. Xu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 510–515 CrossRef CAS PubMed.
- M. J. Leitl, V. A. Krylova, P. I. Djurovich, M. E. Thompson and H. Yersin, J. Am. Chem. Soc., 2014, 136, 16032–16038 CrossRef CAS PubMed.
- M. A. Baldo, C. Adachi and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 10967 CrossRef CAS.
- T. Főrster, Discuss. Faraday Soc., 1959, 27, 7–17 RSC.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS PubMed.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- H. Suzuki and S. Hoshino, J. Appl. Phys., 1996, 79, 8816–8822 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra04929f |
| This journal is © The Royal Society of Chemistry 2015 |