
Yolk–shell nanospheres with soluble amino-polystyrene as a reservoir for Pd NPs†
Guojun Lanab,
Xiaoming Zhangb,
Xiaomin Zhangb,
Mingrun Lib,
Ying Li*a and
Qihua Yang*b
aInstitute of Industrial Catalysis, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: liying@zjut.edu.cn
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China. E-mail: yangqh@dicp.ac.cn
First published on 13th April 2015
Abstract
The fabrication of silica/polymer composites with high polymer content, high stability and unique nanostructures still remains a difficult task though they have wide potential applications in the field of sensing, catalysis, bio-imaging and so on. Herein, the synthesis of yolk–shell nanospheres with soluble amino-polystyrene as a core material (PS-NH2@mesoSiO2 YSNs) have been reported, which is achieved by successive nitration and reduction of polystyrene nanospheres (PS) confined in silica hollow shells. Both the thermal stability and anti-swelling ability of PS-NH2 in the core of yolk–shell nanospheres are greatly enhanced due to the confinement effect. PS-NH2@mesoSiO2 could be used as a reservoir for stabilizing Pd NPs. PS-NH2@mesoSiO2 YSNs with high amino content have high anti-swelling ability and result in Pd NPs with small particle size and high stability. Pd/PS-NH2@mesoSiO2 is an efficient catalyst for the selective hydrogenation of acetophenone (AP) to produce α-phenyl ethanol (PE). NH2 groups in the core of PS-NH2@mesoSiO2 yolk–shell nanospheres not only stabilize Pd nanoparticles but also provide basic surroundings for suppressing the hydrogenolytic splitting of the C–OH to improve the selectivity for α-phenyl ethanol.
Introduction
Metal nanoparticles (NPs) on the borderline of molecular states with discrete quantum energy levels are gaining increasing attention and are considered to bridge the gap between mononuclear metal complexes and heterogeneous bulk catalysts.1 Meanwhile, their large surface area-to-volume ratio and high concentration of low coordination sites and surface vacancies allow effective utilization of noble metals.1b However, being small not only makes the surface atoms dynamically active but also makes the surface of metal NPs unstable, metal NPs often encounter deactivation caused by sintering and/or particle agglomeration during the catalytic reactions.In order to overcome these drawbacks, NPs could be stabilized by capping reagents for the generation of soluble metal NPs.2 Though soluble metal NPs have shown unique catalytic properties, their applications in liquid suspensions are limited due to the difficulties with products separation and catalysts recycling. For facilitating the recycle of the soluble metal NPs, polymer particles have been used as solid support for metal NPs because functional groups similar to those in stabilizing reagents could be incorporated in the polymer.2c,3 For example, spherical polyelectrolyte particles4 and functionalized resins5 have been successfully used for stabilizing metal NPs and metal nanoclusters. However, it is still a rather challenging task for polymer supported metal NPs to ensure the stability of the NPs due to the polymer swelling during the reaction, especially in organic solvents.
Recently, core–shell/yolk–shell nanostructures have been used for hosing metal NPs because the outer shell could isolate metal NPs and prevent their migration and coalescence during the catalytic process.6 Deng and co-workers reported the synthesis of Fe3O4@SiO2–Au@mSiO2 microspheres with high performance in catalytic styrene epoxidation with high conversion and selectivity.7 Liu and co-workers reported the synthesis of yolk–shell structured Au@C nanocomposites that exhibited high catalytic activity in the reduction of 4-nitrophenol.8 Guo and co-workers reported a facile and sacrificial template-free swelling evaporation strategy for the synthesis of Au@polymer yolk–shell nanostructures as efficient heterogeneous catalysts in liquid-phase catalysis.9 However, metal NPs in yolk–shell nanostructures generally have large particle size and the microenvironment of metal NPs is quite different from the soluble metal NPs in most cases. It still lacks efficient method for the synthesis of yolk–shell nanostructures containing metal NPs with ultrafine particle size and retaining their intrinsic catalytic properties.
Herein, we reported the synthesis of yolk–shell nanospheres with soluble amino-polystyrene (PS-NH2) core and mesoporous silica shell as solid stabilizing reagents for metal NPs. Compared with solid polymers, PS-NH2@mesosilica nanostructures have the advantages of (1) the shell could increase the stability of polymer spheres, which in turn may enhance the stability of immobilized metal NPs; (2) the confined nanospace could control the swelling properties of polymer spheres, which may help to tune the exposure degree of functional groups to adjust the catalytic activity of metal NPs; and (3) the enrichment properties of the unique yolk–shell nanostructures may increase the catalytic activity of metal NPs. Our primary results suggest that PS-NH2@mesoSiO2 yolk–shell nanospheres could be used as an efficient and robust solid stabilizing reagent for Pd NPs in the selective hydrogenation of acetophenone to produce α-phenyl ethanol.
Experimental section
Chemicals
All materials were of analytical grade and used as received without any further purification. 1,2-Bis-(trimethoxysilyl) ethane (BTME), cetyltrimethylammonium bromide (CTAB) were purchased from Sigma-Aldrich Company Ltd. (USA). Fluorocarbon surfactant (FC-4) was bought from YickVic Chemicals (Hong Kong). Tetraethoxysilane (TEOS) was obtained from Nanjing Shuguang Chemical Group (China). Other reagents were purchased from Shanghai Chemical Reagent, Inc. of the Chinese Medicine Group. PS@mesoSiO2 YSNs were prepared according to our previous report.10Preparation of PS-NH2@mesoSiO2 yolk–shell nanospheres
PS-NH2@mesoSiO2 yolk–shell nanospheres (YSNs) were prepared by nitration of PS@mesoSiO2 YSNs with HNO3 followed by reduction with SnCl2 with a modified reference method.11 A typical synthetic procedure is described as followings: PS@mesosilicas YSNs (1 g) was added to trifluoroacetic acid (80 mL) at 0 °C. After 10 minutes, 0.5 mL or 5 mL of nitric acid was slowly added and the mixture was stirred for 4 h at 50 °C. The mixture was added to ice/water. After filtration, washing with water, THF, diethylether and drying at 70 °C, a yellow solid was obtained. For reducing the nitro groups, SnCl2·2H2O (10 g) was added to a suspension of PS-NO2@mesosilicas YSNs (1 g) in THF (160 mL) and the mixture was refluxed for 24 h. The solid was filtrated and washed with NaOH (10%), water, THF and diethylether. After drying at 70 °C, PS-NH2@mesoSiO2-S and PS-NH2@mesoSiO2-L were obtained, where S and L refer to 0.5 mL and 5 mL HNO3 used during nitration process, respectively.Synthesis of Pd/PS-NH2@mesoSiO2
Pd/PS-NH2@mesoSiO2 catalysts were prepared by adding an aqueous solution of Na2PdCl4 (20 mL, 0.055 wt%) into aqueous slurry of 0.2 g PS-NH2@mesoSiO2 in 20 mL water. After vigorously stirring at room temperature for 12 h, a solution of NaBH4 in water (10 mL, 0.15 wt%) was added and the resulting suspension was stirred for 6 h at room temperature. Finally, the solid was filtered off, washed with water and ethanol, and dried overnight at 70 °C.Characterizations
The nitrogen sorption experiments were performed at −196 °C using a Micromeritrics ASAP 2020. Samples were degassed at 120 °C for 6 h prior to the measurements. The BET surface area was calculated from the adsorption data in the relative pressure P/P0 range from 0.04 to 0.2. Pore size distributions were determined from the adsorption branches using the nonlocal density functional theory (NLDFT) method. Pore volume was estimated at the relative pressure P/P0 of 0.99. Transmission electron microscopy (TEM) was performed on a HITACHI 7700 at an acceleration voltage of 100 kV. Before the measurement, the sample was dispersed in ethanol and deposited on a holey carbon film on a Cu grid. Scanning transmission electron microscopy (STEM) was undertaken on a HITACHI S-4800 scanning electron microscope operating at an acceleration voltage of 1–20 kV. FT-IR spectra were collected with a Nicolet Nexus 470 IR spectrometer (KBr pellets were prepared) in the range 400–4000 cm−1. The thermo gravimetric analysis (TGA) was performed using a NETZSCH STA 449F3 analyzer from 30 to 900 °C with a heating rate of 10 °C min−1 under air atmosphere.The dispersion of Pd was obtained by CO chemisorption method, which was carried out at 50 °C on a Quantachrome Autosorb-1/C chemisorb apparatus. Prior to measurements, the pre-reduced catalysts were reduced in situ for 2 h at 120 °C in H2. The metal dispersion and particle size were estimated based on the assumption of a spherical geometry of the particles and an adsorption stoichiometry of one CO molecule on one Pd surface atom.12
Titration of amino groups in PS-NH2@mesoSiO2.13 In typical experiment, 50 mg of PS-NH2@mesoSiO2 was added to 6 mL of HCl solution (0.02 M). After stirring at room temperature for 4 h, the mixture was filtrated and the solid was washed with water (50 mL). The obtained filtrate was titrated by a solution of NaOH (5.0 mM).
Catalysis
The following procedure was used for hydrogenation of acetophenone: Pd/PS-NH2@mesoSiO2 catalysts (0.00375 mmol of Pd) were added into a reaction glass vial fitted with a magnetic stirring bar and a septum cap, followed by the acetophenone (0.75 mmol) in 2 mL ethanol. The reaction vial was then placed into a 300 mL steel Parr autoclave. The autoclave was flushed with hydrogen twice and then it was pressurized to 4 bar hydrogen pressure and placed into water bath at 60 °C. After the reaction, the autoclave was placed into a water bath and cooled to room temperature. Finally, the remaining hydrogen gas was discharged and the products were analyzed by an Agilent 6890 GC equipped with a HP-5 capillary column with FID detector.Results and discussion
Synthesis and characterization of PS-NH2@mesoSiO2 YSNs
According to our previous report, PS@mesoSiO2 YSNs with particle size of 300 nm and shell thickness of 50 nm were synthesized by wrapping PS NPs with TEOS followed by the addition of BTME in base medium with CTAB as structure directing agents10 (Fig. S1†). PS-NH2@mesoSiO2 was obtained by nitration and reduction of PS@mesoSiO2 YSNs. The content of NO2 groups incorporated onto PS@mesoSiO2 YSNs could be controlled by the amount of HNO3 during the nitration process. The successive reduction could directly result in the formation of PS-NH2@mesoSiO2-S and PS-NH2@mesoSiO2-L YSNs, where S and L refers lower and higher content of HNO3 used during nitration process. The TEM images show that both PS-NH2@mesoSiO2-S and PS-NH2@mesoSiO2-L have yolk–shell nanostructure with particle size of 300 nm, shell thickness of 50 nm and core size of 180 nm similar to parent PS@mesoSiO2 YSNs, showing that the yolk–shell nanostructure is quite stable to survive the nitration and reduction process (Fig. 1). | ||
| Fig. 1 TEM images of (A) PS-NH2@mesoSiO2-S and (B) PS-NH2@mesoSiO2-L: (a) fresh sample (b) after treatment with toluene and (c) after treatment with TEA/toluene = 1. | ||
The FT-IR spectra of PS@mesoSiO2 YSNs during the nitration and reduction process are summarized in Fig. 2. The FT-IR spectrum of PS@mesoSiO2 YSNs displays the typical vibration peaks for aromatic ring of PS around 1450–1630 cm−1, the stretch vibrations of the C–H bond of the bridging –CH2CH2– in the network of silica shell and –CH2– of PS around 2950–2840 cm−1. After nitration with nitric acid in trifluoroacetic acid, the typical vibration peaks corresponding to nitro group appear at 1529 and 1348 cm−1, showing the incorporation of NO2 groups in aromatic rings of PS. After reduction of the nitro groups with solid SnCl2·2H2O in THF, the vibration peaks for NO2 become weak and a new vibration peak at 1513 cm−1 assigned to the in-plane bending vibration of NH2 groups14 appears, showing that most NO2 groups have been successfully transformed to NH2 during reduction process.11 The existence of NO2 after reduction is probably due to the fact that some NO2 group in PS cannot be accessed by the reductant. PS-NH2@mesoSiO2-S YSNs and PS-NH2@mesoSiO2-L YSNs have almost similar FT-IR spectrum, showing the successfully incorporation of NH2 groups in spite of the amount of nitric acid used during the nitration process (Fig. S2†).
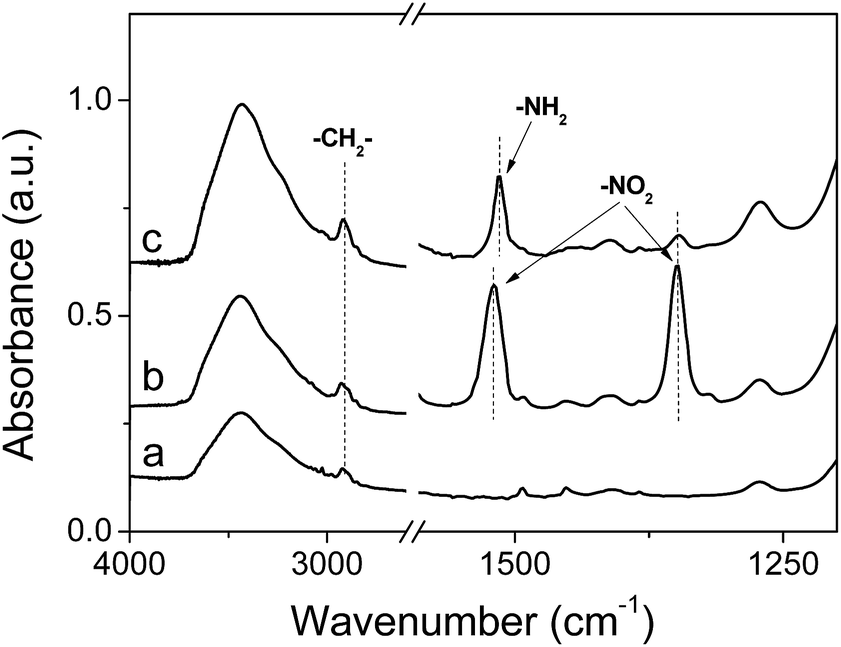 | ||
| Fig. 2 IR spectra of (a) PS@mesoSiO2 YSNs, (b) PS@mesoSiO2 after nitration with higher amount of HNO3 (5 mL) and (c) PS-NH2@mesoSiO2-L YSNs. | ||
Elemental analysis shows that PS-NH2@mesoSiO2-L has higher N content than PS-NH2@mesoSiO2-S, suggesting that higher amount of nitric acid could result in higher nitration degree of PS (Table 1). Based on the TG analysis, the polymer content for PS-NH2@mesoSiO2-S and PS-NH2@mesoSiO2-L is quite similar (36.2 versus 40.0 wt%) (Table 1). The combined results of TG and elemental analysis show that 0.22 units of N could be incorporated in each unit of aromatic ring of PS for PS-NH2@mesoSiO2-S and PS-NO2@mesoSiO2-L could have 0.69 units of nitro groups incorporated to each unit of aromatic ring of PS. The titration experiment shows that base exchange capacity for PS-NH2@mesoSiO2-S and PS-NH2@mesoSiO2-L is 0.30 and 0.54 mmol g−1, respectively. The lower NH2 content than N content is either due to the incomplete reduction of NO2 or inaccessibility of some NH2 during the titration process.
| Sample | Polymer contenta (wt%) | Nb (mmol g−1) | NH2c (mmol g−1) | SBET (m2 g−1) | Pore volume (cm3 g−1) | Pore diameter (nm) | CO2 capacityd (mmol g−1) | |
|---|---|---|---|---|---|---|---|---|
| 0 °C | 25 °C | |||||||
| a Obtained from weight loss in the range of 200–700 °C based on TG analysis.b Measured by C, H, N elemental analysis.c Measured by titration.d The CO2 adsorption is performed under pressure of 0–760 mm Hg. | ||||||||
| PS-NH2@mesoSiO2-S | 36.2 | 0.72 | 0.30 | 565 | 0.47 | 1.3/2.4 | 1.10 | 0.71 |
| PS-NH2@mesoSiO2-L | 40.0 | 2.35 | 0.54 | 499 | 0.44 | 1.3/2.4 | 1.16 | 0.97 |
The amount of basic surface site in the PS-NH2@mesoSiO2 YSNs was also estimated based on the CO2 adsorption capacities. The adsorption isotherms of CO2 on PS-NH2@mesoSiO2 YSNs at 25 °C under pressure of 0–760 mm Hg are shown in Fig. 3A. The isotherms of CO2 on PS-NH2@mesoSiO2 YSNs are of type I and a sharp increase in CO2 adsorption amount was observed at pressure range from about 0 to 20 mm Hg. This suggests the chemical adsorption behavior between CO2 and adsorbents,15–17 further confirming the existence of NH2 groups on PS-NH2@mesoSiO2 YSNs. PS-NH2@mesoSiO2-S and PS-NH2@mesoSiO2-L exhibit CO2 adsorption capacity of 0.71 mmol g−1 and 0.97 mmol g−1 under the pressure of 760 mm Hg, respectively. CO2 adsorption amount is higher on PS-NH2@mesoSiO2-L than on PS-NH2@mesoSiO2-S, in spite of the fact that the BET surface area and pore volume of PS-NH2@mesoSiO2-L are lower than those of PS-NH2@mesoSiO2-S (Table 1).
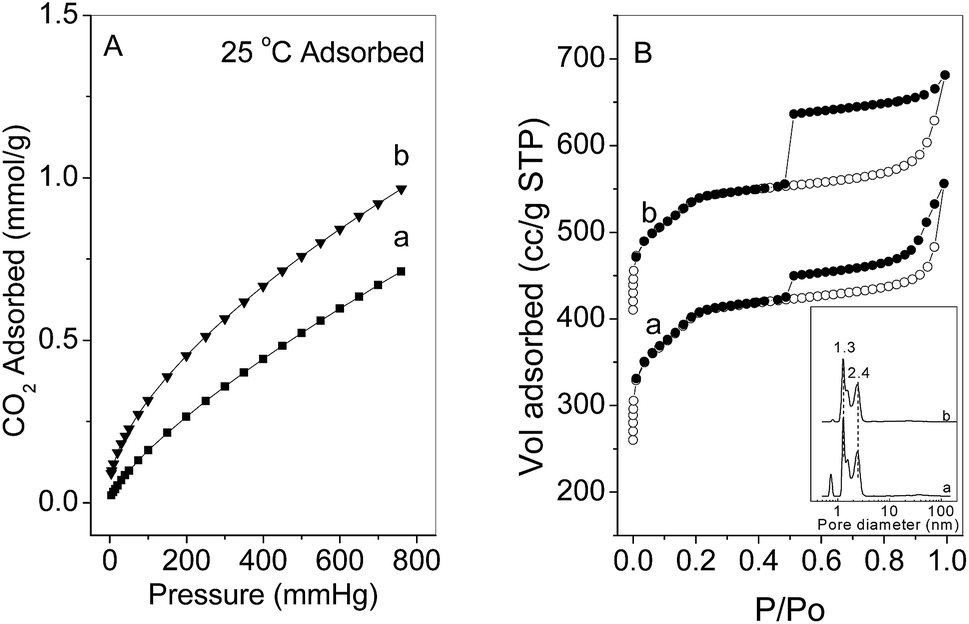 | ||
| Fig. 3 (A) CO2 adsorption isotherms at 25 °C under pressure of 0–760 mm Hg and (B) N2 sorption isotherms for (a) PS-NH2@mesoSiO2-S and (b) PS-NH2@mesoSiO2-L. | ||
The nitrogen adsorption–desorption isotherms of PS-NH2@mesoSiO2 YSNs display typical IV isotherm pattern with a sharp capillary condensation step and H4-type hysteresis loop in the P/P0 range of 0.5–1.0 (Fig. 3B). This indicates the presence of mesopores in the outer shell according to IUPAC classification. The detailed pore size distribution calculated based on the NLDFT reveals that the PS-NH2@mesoSiO2 YSNs have uniform pores of about 1.3 nm and 2.4 nm. The existence of the mesopores in the outer shell will benefit the free diffusion of guest molecules throughout the nanospheres. It should be mentioned that the H4 hysteresis in the N2 sorption isotherm of PS-NH2@mesoSiO2-L is probably from the hollow interiors of the PS-NH2@mesoSiO2. PS-NH2@mesoSiO2-L has slightly lower BET surface area and pore volume than PS-NH2@mesoSiO2-S (Table 1).
Stability of PS-NH2@mesoSiO2 YSNs
The TG curves of PS-NH2 (obtained by etching SiO2 shell of PS-NH2@mesoSiO2-L) (Fig. 4), and PS-NH2@mesoSiO2 samples exhibit three consecutive weight loss steps in the range of 30 to 700 °C, which is due to the loss of the physically adsorbed water, decomposition of NH2 groups, and destruction of polymer framework. For PS-NH2@mesoSiO2 samples, the decomposition of ethane-silica in the shell takes place at temperature above 400 °C.10 The decomposition temperature of PS-NH2@mesoSiO2 samples is much higher than that of PS-NH2 (400 °C versus 200 °C), probably due to the protective effect of the shell. The more distinctive weight loss step for PS-NH2@mesoSiO2-S than PS-NH2@mesoSiO2-L is due to the lower N content.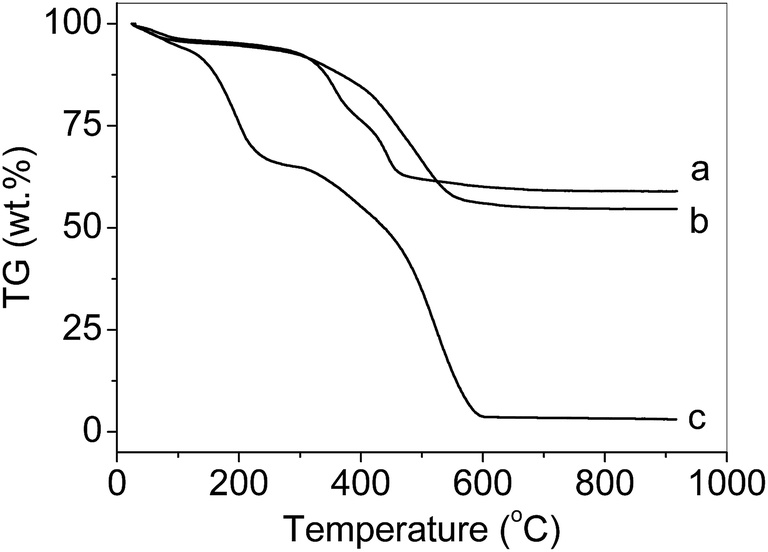 | ||
| Fig. 4 TG curves of (a) PS-NH2@mesoSiO2-S, (b) PS-NH2@mesoSiO2-L and (c) PS-NH2 under the air. | ||
The stability of PS-NH2@mesoSiO2 samples in the organic solvent is an important issue for their application as solid stabilizing reagent for metal NPs. The TEM images of PS-NH2@mesoSiO2 samples after treatment in organic solvent are displayed in Fig. 1 and S3.† After treatment in toluene and in mixed solvent of triethylamine (TEA)/toluene with volume ratio of 1, PS-NH2 swells immediately and no solid could be observed. For PS-NH2@mesoSiO2-S, the core becomes illegible and shell becomes thicker (50 versus 70 nm) after treatment in toluene. This suggests that PS-NH2 core swells and the silica shell prevents the swelled PS-NH2 to leak out of the yolk–shell nanospheres due to the H bond and hydrophobic interactions among PS-NH2 and silica shell. After treatment in TEA/toluene, the yolk–shell nanospheres transfers to double-shell hollow nanospheres. The TG analysis shows that the weight loss of PS-NH2@mesoSiO2-S before and after treatment remains almost the same, showing no leakage of PS-NH2 occurs during the treatment process (Fig. S4†). Interestingly, no obvious change in morphology and no volume expansion of the core could be observed for PS-NH2@mesoSiO2-L. This suggests that PS-NH2@mesoSiO2-L has higher anti-swelling ability than PS-NH2@mesoSiO2-S, probably due to the existence of higher content of NH2 in the former sample.
In a confined nanospace, PS-NH2 is only surrounded by small amount of solvent due to the limited space volume. Thus, the competition of swelling and shrinkage of PS-NH2 (driven by the H–H bond interactions among NH2 group) may occur. The solvent amount in the confined nanospace remains almost the same, therefore, the volume expansion and shrinkage of PS-NH2 is mainly determined by the strength of H–H bond interactions among NH2 group. PS-NH2@mesoSiO2-S with less amount of NH2 moiety has weaker H–H bond interactions among NH2 group. Therefore, it swells obviously after treatment in toluene as observed in Fig. 1A(b). The addition of TEA in toluene may enhance the H–H bond interactions among NH2 group. The volume decrease of PS-NH2 from ca. 3.1 × 106 nm3 to 2.4 × 106 nm3 for PS-NH2@mesoSiO2-S after treatment with toluene/TEA further confirms the enhanced H–H bond interactions among NH2 group. As a result, the double shell nanostructure was formed. The similar phenomenon was also observed by confining PS-SO3H in a confined nanospace in our previous work.10
Compared with PS-NH2, PS-NH2@mesoSiO2 YSNs exhibits less swelling ability, which is likely due to the confinement effect of the yolk–shell nanospheres. The PS-NH2@mesoSiO2-L also shows high stability in various polar solvents such as ethyl acetate, dioxane, acetonitrile, N,N-dimethyl amide, thionyl chloride and dichloromethane (Fig. S3†). The higher stability of PS-NH2@mesoSiO2-L with higher amount of PS-NH2 is mainly attributed to the stronger H–H bond interactions among NH2 group.
PS-NH2@mesoSiO2 YSNs as solid stabilizing reagents for Pd NPs
Pd NPs could be deposited in PS-NH2@mesoSiO2 YSNs for the formation of Pd/PS-NH2@mesoSiO2 YSNs via simple adsorption followed by reduction with Na2PdCl4 as a metal source. Through adjusting the content of Na2PdCl4, the loading amount of Pd could be varied. The STEM images of 1wt%Pd/PS-NH2@mesoSiO2-L and 2wt%Pd/PS-NH2@mesoSiO2-L shown in Fig. 5 reveal that Pd particles with narrow particle size distribution are finely and uniformly dispersed in the core of PS-NH2@mesoSiO2 YSNs. The particle size of Pd NPs for 1wt%Pd/PS-NH2@mesoSiO2-L and 2wt%Pd/PS-NH2@mesoSiO2-L is in the range of 2.0–3.0 nm and 6.0–7.0 nm, respectively. As control samples, PS-NH2@mesoSiO2-S, was also used as support for Pd NPs. The STEM images show that Pd particles of 2wt%Pd/PS-NH2@mesoSiO2-S have wider particle size distribution and larger particle size than that of 2wt%Pd/PS-NH2@mesoSiO2-L. This indicates that NH2 group acts as anchoring sites for Na2PdCl4 and PS-NH2@mesoSiO2 YSNs could be regarded as solid stabilizing reagents for Pd NPs. The particle size of Pd NPs could be simply adjusted by varying Pd content and NH2 content in PS-NH2.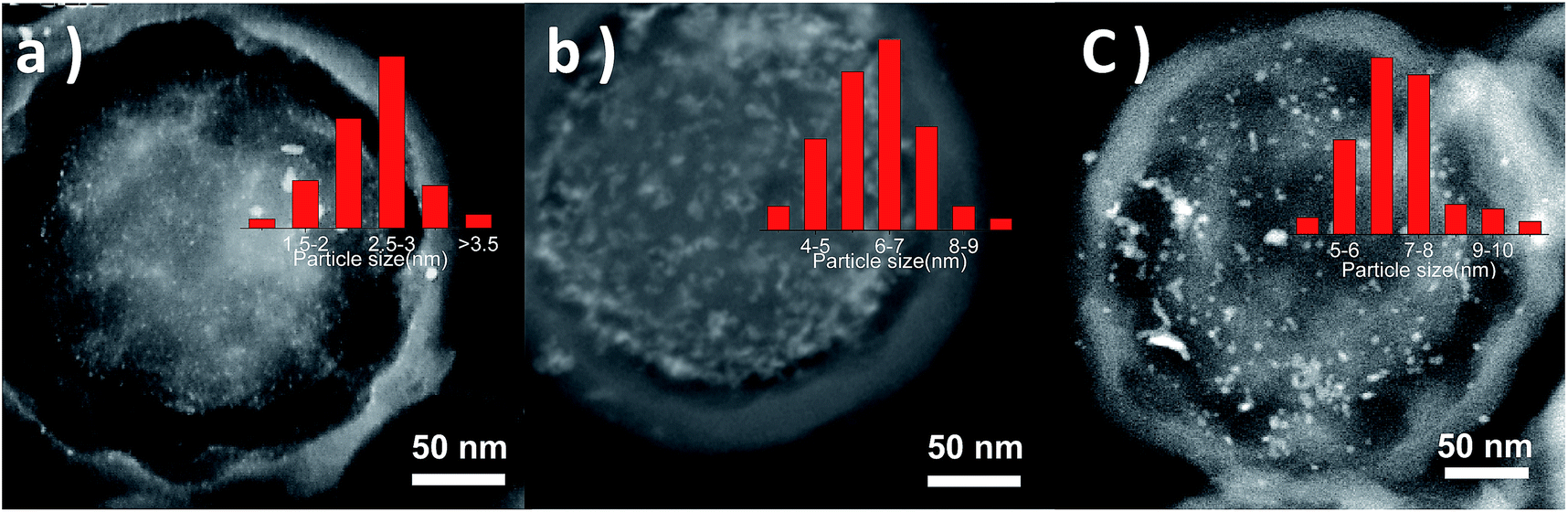 | ||
| Fig. 5 STEM images of (a) 1 wt% Pd/PS-NH2@mesoSiO2-L, (b) 2wt%Pd/PS-NH2@mesoSiO2-L, and (c) 2wt%Pd/PS-NH2@mesoSiO2-S (inset is the particle size distribution for Pd NPs). | ||
CO chemisorption was also performed for characterizing the distribution of Pd on PS-NH2@mesoSiO2 YSNs samples (Table 2). The dispersion of Pd increases in the order of 1wt%Pd/PS-NH2@mesoSiO2-L > 2wt%Pd/PS-NH2@mesoSiO2-L > 2wt%Pd/PS-NH2@mesoSiO2-S. The particle size of Pd based on chemisorption method is quite similar to the results of STEM characterizations, further confirming that higher content of NH2 group in PS-NH2@mesoSiO2 benefits higher dispersion degree of Pd NPs. As shown in Tables 1 and S1,† the pore diameter of PS-NH2@mesoSiO2 YSNs before and after Pd loading remains almost the same. Only slight decrease in BET surface area (540 to 521 m2 g−1) and pore volume (0.45 to 0.37 cm3 g−1) could be observed after Pd loading, probably due to the dissolution of silica residues during NaBH4 reduction process.
| Pd catalysts | CO chemisorptions | Conv. (%) | Sela (%) | TOF (h−1) | ||
|---|---|---|---|---|---|---|
| Pd surface area (m2 g−1) | Pd particles size (nm) | Dispersion (%) | ||||
| a Selectivity to α-phenyl ethanol, reaction conditions: S/C = 200, solvent (2 mL ethanol), P = 4 bar (H2), T = 60 °C, reaction time, 1 h. TOF was calculated with conversion below 30%, the conversion and selectivity were determined by GC.b From literature data.19c From literature data.20 | ||||||
| 2wt%Pd/PS-NH2@mesoSiO2-S | 0.88 | 11.3 | 9.9 | 53.0 | 98.1 | 1182 |
| 2wt%Pd/PS-NH2@mesoSiO2-L | 1.52 | 6.6 | 17.1 | 82.8 | 98.9 | 690 |
| 1wt%Pd/PS-NH2@mesoSiO2-L | 1.67 | 3.0 | 37.5 | 99.0 | 98.6 | 1072 |
| Pd/ACb | — | — | — | 100 | 60 | — |
| Pd/ACc | — | — | — | ∼80 | ∼60 | |
Selective hydrogenation of acetophenone
The experimental results of the selective hydrogenation of acetophenone (AP) over different catalysts are shown in Table 2. Selective hydrogenation of AP to α-phenylethanol (PE) is complicated by the fact that different side reactions may take place. The general reaction network, with all the possible reaction pathways during AP hydrogenation, is depicted in Scheme 1. Hydrogenation of carbonyl group of AP gives PE, whereas the hydrogenation of the aromatic ring leads to cyclohexylmethylketone (CHMK). Both primary products, PE and CHMK, can subsequently be hydrogenated to 1-cyclohexylethanol (CHE). The hydrogenolytic splitting of the C–OH bond of PE produces ethylbenzene (EB). In consequence, achieving high selectivity to α-phenylethanol, the desired product, is a challenging issue.18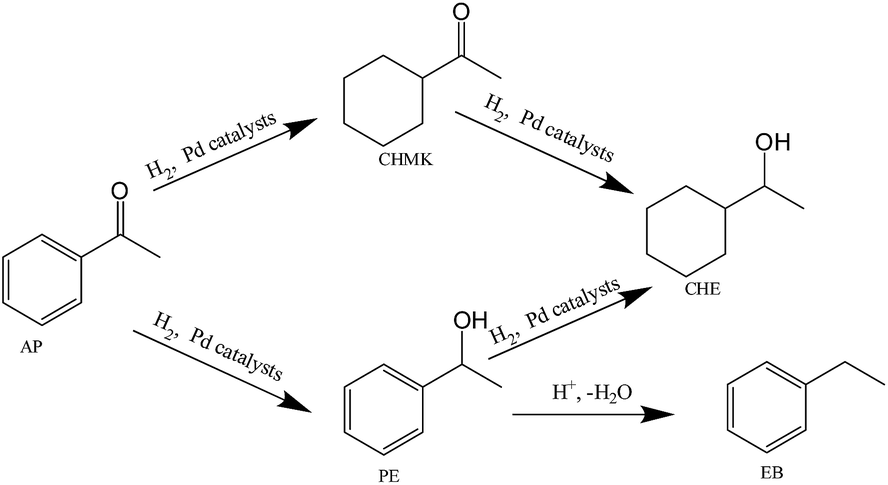 | ||
| Scheme 1 Reaction network for acetophenone hydrogenation over Pd catalysts. | ||
The selective hydrogenation of acetophenone to α-phenyl ethanol is performed at mild reaction conditions at 60 °C with H2 pressure of 4 bar. All Pd supported on PS-NH2@mesoSiO2 samples could efficiently catalyze the acetophenone hydrogenation with α-phenyl ethanol (PE) as main product. The previous studies18–21 show that Pd supported on active carbon (AC) affords ethyl benzene as the major product due to the acidic properties of AC (typically Pd/AC catalysts exhibit acidic surface groups, such as carbonyl, carboxylic, phenolic hydroxyl, lactone and quinone groups). The high selectivity of Pd/PS-NH2@mesoSiO2 to PE is mainly attributed to the presence of basic NH2 which suppresses hydrogenolytic splitting of the C–OH bond of PE. This suggests that the NH2 groups not only play important role in stabilizing Pd nanoparticles but also improve the selectivity for α-phenyl ethanol.
1wt%Pd/PS-NH2@mesoSiO2-L shows higher TOF than 2wt%Pd/PS-NH2@mesoSiO2-L (1072 versus 690 h−1), suggesting that Pd with small particle size has higher activity. However, 2wt%Pd/PS-NH2@mesoSiO2-S with the largest Pd NPs shows the highest TOF among all the samples tested. The titration experiments show that PS-NH2@mesoSiO2-L has more NH2 groups than PS-NH2@mesoSiO2-S. The lower activity of the former sample implies that NH2 group could deactivate the Pd NPs though they could also stabilize Pd NPs.
The stability of the solid catalyst was investigated using 1wt%Pd/PS-NH2@mesoSiO2-L and 2wt%Pd/PS-NH2@mesoSiO2-L as model catalysts in the hydrogenation of acetophenone (Fig. 6). Almost no conversion could be observed for the second cycle on 2wt%Pd/PS-NH2@mesoSiO2-L. Interestingly, 1wt%Pd/PS-NH2@mesoSiO2-L could be stably reused for more than 10 times without obvious loss of activity and selectivity. The TEM images of 1wt%Pd/PS-NH2@mesoSiO2-L after 10 cycles and 2wt%Pd/PS-NH2@mesoSiO2-L after 1 cycle are shown in Fig. 7 and S5,† respectively. There is no obvious change in the yolk–shell nanostructure for both the above two catalysts, showing that the yolk–shell nanostructure is robust enough during the catalysis. Severe aggregation of Pd NPs was observed in the TEM image of reused 2wt%Pd/PS-NH2@mesoSiO2-L. On the contrary, most Pd particles in 1wt%Pd/PS-NH2@mesoSiO2-L after 10 cycles still have small size centered at 3–4 nm and only small amount of large aggregates of Pd particles could also be observed. The different recycle behavior of 1wt%Pd/PS-NH2@mesoSiO2-L and 2wt%Pd/PS-NH2@mesoSiO2-L suggests that higher NH2/Pd ratio benefits the stability of Pd NPs during the recycle process.
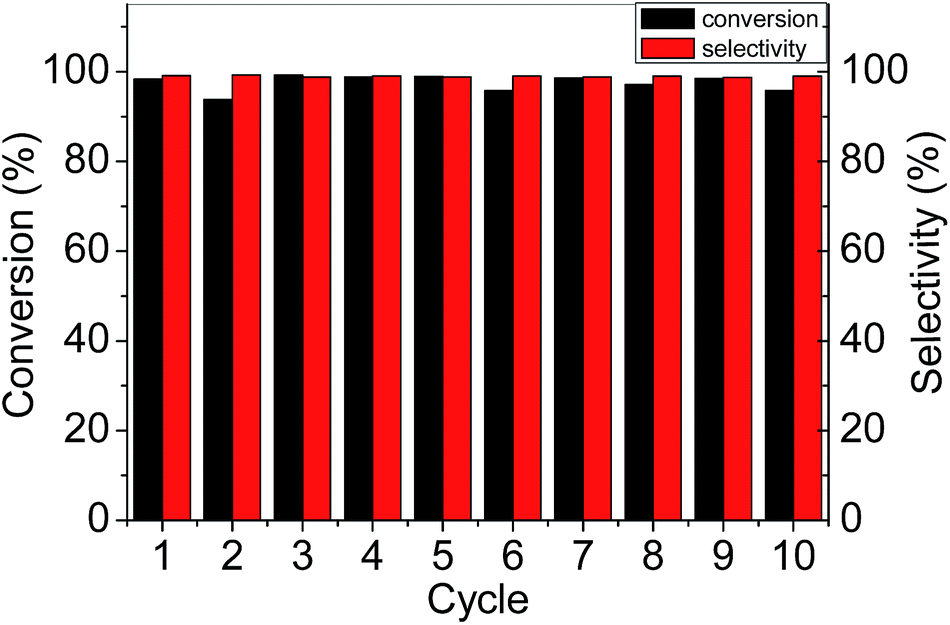 | ||
| Fig. 6 Reusability of 1wt%Pd/PS-NH2@mesoSiO2-L in hydrogenation of acetophenone. | ||
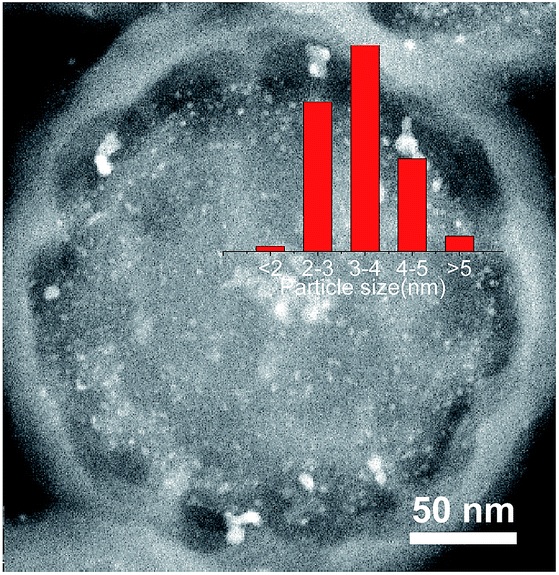 | ||
| Fig. 7 STEM image of 1wt%Pd/PS-NH2@mesoSiO2-L after 10 cycles. | ||
Conclusions
In summary, PS-NH2@mesoSiO2 were prepared by nitration and reduction of polystyrene nanospheres confined in silica hollow shell. The content of amine group could be adjusted by the amount of HNO3 used in the nitration process. It was found that PS-NH2@mesoSiO2 with higher content of amine groups show higher anti-swelling ability and could result in the formation of smaller Pd NPs. Pd/PS-NH2@mesoSiO2 YSNs could efficiently catalyze the selective hydrogenation of acetophenone to produce phenyl ethanol. The high selectivity to phenyl ethanol is attributed to the presence of amine groups which could suppress the hydrogenolytic splitting of the C–OH. The primary result also suggests that high content of amine group could have high stabilizing effect for Pd NPs with sacrifice of the activity.Acknowledgements
This work was financially supported by Natural Science Foundation of China (21325313, 21321002) and by the Key Research Program of the Chinese Academy of Sciences (Grant no. KGZD-EW-T05).Notes and references
- (a) B. R. Cuenya, Acc. Chem. Res., 2013, 46, 1682–1691 CrossRef PubMed; (b) A. Cao, R. Lu and G. Veser, Phys. Chem. Chem. Phys., 2010, 12, 13499–13510 RSC; (c) Y. N. Xia, Y. J. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed; (d) K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1512–1524 CrossRef CAS PubMed.
- (a) A. Gual, C. Godard, S. Castillon, D. Curulla-Ferre and C. Claver, Catal. Today, 2012, 183, 154–171 CrossRef CAS PubMed; (b) N. Yan, C. X. Xiao and Y. Kou, Coord. Chem. Rev., 2010, 254, 1179–1218 CrossRef CAS PubMed; (c) S. Mahouche-Chergui, M. Guerrouache, B. Carbonnier and M. M. Chehimi, Colloids Surf., A, 2013, 439, 43–68 CrossRef CAS PubMed.
- N. D. Golubeva, B. K. Dyusenalin, B. S. Selenova, S. I. Pomogailo, A. K. Zharmagambetova, G. I. Dzhardimalieva and A. D. Pomogailo, Kinet. Catal., 2011, 52, 242–250 CrossRef CAS.
- Y. Mei, Y. Lu, F. Polzer, M. Ballauff and M. Drechsler, Chem. Mater., 2007, 19, 1062–1069 CrossRef CAS.
- (a) T. Teranishi, K. Nakata, M. Iwamoto, M. Miyake and N. Toshima, React. Funct. Polym., 1998, 37, 111–119 CrossRef CAS; (b) S. K. Mahato, R. U. Islam, C. Acharya, M. J. Witcomb and K. Mallick, ChemCatChem, 2014, 6, 1419–1426 CAS; (c) L. Y. Li, H. X. Zhao, J. Y. Wang and R. H. Wang, ACS Nano, 2014, 8, 5352–5364 CrossRef CAS PubMed.
- (a) Q. Zhang, I. Lee, J. B. Joo, F. Zaera and Y. D. Yin, Acc. Chem. Res., 2013, 46, 1816–1824 CrossRef CAS PubMed; (b) J. Liu, H. Q. Yang, F. Kleitz, Z. G. Chen, T. Y. Yang, E. Strounina, G. Q. Lu and S. Z. Qiao, Adv. Funct. Mater., 2013, 22, 591–599 CrossRef PubMed; (c) S. J. Peng, L. L. Li, H. T. Tan, R. Cai, W. H. Shi, C. C. Li, S. G. Mhaisalkar, M. Srinivasan, S. Ramakrishna and Q. Y. Yan, Adv. Funct. Mater., 2014, 24, 2155–2162 CrossRef CAS PubMed.
- Y. H. Deng, Y. Cai, Z. K. Sun, J. Liu, C. Liu, J. Wei, W. Li, C. Liu, Y. Wang and D. Y. Zhao, J. Am. Chem. Soc., 2010, 132, 8466–8473 CrossRef CAS PubMed.
- R. Liu, S. M. Mahurin, C. Li, R. R. Unocic, J. C. Idrobo, H. J. Gao, S. J. Pennycook and S. Dai, Angew. Chem., Int. Ed., 2011, 50, 6799–6802 CrossRef CAS PubMed.
- J. Han, R. Chen, M. G. Wang, S. Lu and R. Guo, Chem. Commun., 2013, 49, 11566–11568 RSC.
- X. M. Zhang, Y. P. Zhao, S. T. Xu, Y. Yang, J. Liu, Y. X. Wei and Q. H. Yang, Nat. Commun., 2014, 5, 310 Search PubMed.
- E. Merino, E. Verde-Sesto, E. M. Maya, M. Iglesias, F. Sanchez and A. Corma, Chem. Mater., 2013, 25, 981–988 CrossRef CAS.
- G. Agostini, R. Pellegrini, G. Leofanti, L. Bertinetti, S. Bertarione, E. Groppo, A. Zecchina and C. Lamberti, J. Phys. Chem. C, 2009, 113, 10485–10492 CAS.
- J. Alauzun, A. Mehdi, C. Reye and R. J. P. Corriu, J. Am. Chem. Soc., 2006, 128, 8718–8719 CrossRef CAS PubMed.
- Y. Yao, H. Xiao, P. Wang, P. P. Su, Z. G. Shao and Q. H. Yang, J. Mater. Chem. A, 2014, 2, 11768–11775 CAS.
- S. Y. Bai, J. Liu, J. S. Gao, Q. H. Yang and C. Li, Microporous Mesoporous Mater., 2012, 151, 474–480 CrossRef CAS PubMed.
- Y. Belmabkhout and A. Sayari, Adsorption, 2009, 15, 318–328 CrossRef CAS PubMed.
- M. Cristina and F. Tomislav, Angew. Chem., Int. Ed., 2014, 53, 7471–7474 CrossRef PubMed.
- (a) P. Maki-Arvela, S. Sahin, N. Kumar, J. P. Mikkola, K. Eranen, T. Salmi and D. Y. Murzin, Catal. Today, 2009, 140, 70–73 CrossRef PubMed; (b) S. Sahin, P. Maki-Arvela, J. P. Tessonnier, A. Villa, S. Reiche, S. Wrabetz, D. S. Su, R. Schlogl, T. Salmi and D. Y. Murzin, Appl. Catal., A, 2011, 408, 137–147 CrossRef CAS PubMed; (c) C. S. Chen and H. W. Chen, Appl. Catal., A, 2004, 260, 207–213 CrossRef CAS PubMed.
- M. Lenarda, M. Casagrande, E. Moretti, L. Storaro, R. Frattini and S. Polizzi, Catal. Lett., 2007, 114, 79–84 CrossRef CAS.
- Y. Z. Xiang, Y. A. Lv, T. Y. Xu, X. N. Li and J. G. Wang, J. Mol. Catal. A: Chem., 2011, 351, 70–75 CrossRef CAS PubMed.
- P. Maki-Arvela, S. Sahin, N. Kumar, T. Heikkila, V. P. Lehto, T. Salmi and D. Y. Murzin, J. Mol. Catal. A: Chem., 2008, 285, 132–141 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S5 and Table S1. See DOI: 10.1039/c5ra04923g |
| This journal is © The Royal Society of Chemistry 2015 |