
Reverse iodine transfer polymerization (RITP) of chloroprene†
Abstract
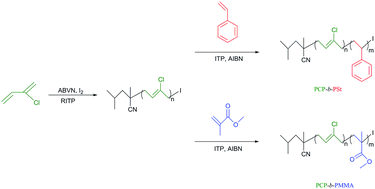
Reversible-deactivation radical polymerization (RDRP) of chloroprene (2-chloro-1,3-butadiene, CP) using reverse iodine transfer polymerization (RITP) has been demonstrated for the first time. Reverse iodine transfer polymerizations of CP were studied in benzene at 50 °C using 2,2′-azobis(isoheptonitrile) (ABVN) as a radical initiator, where a molar ratio ABVN/I2 = 1.7 was used. The process efficiently controls the molar mass (characterized by size exclusion chromatography, SEC) and the structure of the polymer (confirmed by 1H NMR spectra). For example, polychloroprene (PCP) samples of Mn,SEC = 4900 g mol−1 and Mw/Mn = 1.73 (Mn,th = 4800 g mol−1), Mn,SEC = 7300 g mol−1 and Mw/Mn = 1.89 (Mn,th = 7300 g mol−1), and Mn,SEC = 9000 g mol−1 and Mw/Mn = 1.86 (Mn,th = 9300 g mol−1) were successfully obtained. Furthermore, the influence of solvent, initiator and temperature were studied. Last, the controlled nature of the polymer was confirmed by the preparation of polychloroprene-b-polystyrene (PCP-b-PSt) and polychloroprene-b-poly(methyl methacrylate) (PCP-b-PMMA) diblock copolymers.
 Please wait while we load your content...
Please wait while we load your content...

