
Coordination-driven self-assembly: construction of a Fe3O4–graphene hybrid 3D framework and its long cycle lifetime for lithium-ion batteries†
Zhimin Ren,
Siqi Yu,
Xinxin Fu,
Lin Shi,
Chunxiao Sun,
Chenyao Fan,
Qi Liu,
Guodong Qian and
Zhiyu Wang*
State Key Laboratory of Silicon Materials, Department of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: wangzhiyu@zju.edu.cn; Fax: +86-571-87952539; Tel: +86-571-87952539
First published on 13th April 2015
Abstract
In this study, we have demonstrated coordination-driven self-assembly between Fe3O4 and graphene sheets under a hydrothermal condition for the simple in situ synthesis of a 3D Fe3O4–graphene hybrid architecture (Fe3O4/G). Fine hierarchical Fe3O4 spheres were homogeneously dispersed and embedded in an interconnected mesoporous framework of graphene sheets. It can be noted that the critical concentration of GO assembly decreased dramatically during the self-assembly of Fe3O4, indicating the coordination-driven self-assembly between Fe3O4 and GO. When evaluated as an anode material for LIBs, the Fe3O4/G hybrid framework demonstrates a high reversible capacity of 1164 mA h g−1 over 500 cycles at a current density of 500 mA g−1 and a remarkable rate capability. The superior electrochemical performance is attributed to a strong adhesion force and synergistic effect between Fe3O4 and graphene sheets as well as formation of a 3D interconnected hybrid framework, which offers enhanced kinetics towards electrochemical reactions with lithium ions and provides space for alleviating the huge volume expansion that occurs during charge–discharge cycles.
Introduction
One of the factors that limit the energy density of lithium ion batteries (LIBs) is the carbon-based anode materials like graphite, which have a low specific capacity of 372 mA h g−1. Many efforts have been made toward developing new electrode materials with high energy densities to satisfy the ever-growing demand for high performance LIBs.1–6 Currently, transition metal oxide (CuO, Co2O3, NiO, and Fe3O4) has attracted increasing attention because of many advantages such as safety, nontoxicity and high theoretical capacities.7–11 Among them, Fe3O4 is regarded as a promising anode material to replace graphite electrodes due to its low cost, eco-friendliness, natural abundance, and relatively high electronic conductivity compared with other metal oxides.12–15 Fe3O4 electrodes are based on the “conversion reaction” in the charge–discharge cycles, resulting in a large volume expansion (>200%) and poor capacity retention, which greatly hinder the practical utilization of Fe3O4 anodes.5,16–18Downsizing particles to the nanoscale with special morphologies is an effective way to improve the electrochemical properties because of the high surface areas, large pore volumes and short diffusion lengths for both Li ions and electrons. In particular, the hierarchical micro-structure constructed by nanoparticles has aroused tremendous interest due to the combined advantages of nano- and micro-materials.19–21 Another feasible approach is to form hybrid materials by introducing conducting carbon matrices to absorb the volume changes and improve the electronic conductivity of the electrode.22–24 Graphene, a two-dimensional single layer sheet of hexagonal carbon, has emerged as one of the most appealing carbon matrices for metal oxide particles because of its excellent electrical conductivity, high surface area, and good chemical and thermal stability. Graphene oxide (GO), which has abundant functional groups and exhibits strong adhesion to metal ions, is often used in situ for the preparation of nanoparticles/graphene composites.25,26 Recent works have revealed that 2D graphene oxide assembles into 3D architectures under hydrothermal conditions if the concentration of GO is at least 1 mg mL−1.27 Using a similar method, a Fe3O4–graphene hybrid framework can also be prepared by introducing Fe3O4 nanocrystals or Fe3+ into a GO suspension (GO concentration is up to 1.5 mg mL−1). With this in mind, numerous GO-based Fe3O4 host materials have been successfully synthesized and show enhanced properties.28–32 However, most of the studies focused on the assembly of graphene oxide. There are still very few reports on the simultaneous co-assembly process of Fe3O4 nanocrystals and graphene oxide.
In this study, we prepared a three-dimensional graphene framework with a uniform distribution of hierarchical Fe3O4 spheres via a one-pot solvothermal method. Accompanied with the formation and assembly of Fe3O4 crystals, GO is also reduced and assembled into a 3D network at the same time. The two assembly processes reinforced and interacted with each other through coordination-driven self-assembly to form the integral Fe3O4–graphene hybrid framework (Fe3O4/G). The critical feature of this method is the usage of an ethylene glycol–water system and sodium oleate. The as-formed 3D hybrid framework has the advantage of the two building blocks and exhibits an outstanding reversible capacity and excellent rate performance.
Materials and methods
Materials
Sodium oleate (96%, Aladdin), iron(III) chloride (FeCl3·6H2O, chemically pure, Sinopharm Chemical Reagent), ethylene glycol (chemically pure, Sinopharm Chemical Reagent), iron(III) sulfate hydrate, (Fe2(SO4)3·xH2O, chemically pure, Sinopharm Chemical Reagent), graphene oxide solution (2 mg mL−1, Sinocarbon Graphene Marketing Center, Chinese Academy of Science), and ethanol (chemically pure, Sinopharm Chemical Reagent) were used as received without further purification.Synthesis
Fe3O4/G was prepared by a one-pot solvothermal method. In a typical synthesis process, 5 mmol of sodium oleate was dissolved in 30 mL of the mixed solution (ethylene glycol–graphene oxide solution) by vigorous stirring for 1 h, followed by adding 3 mmol of FeCl3·6H2O to form a uniform gray dispersion. Subsequently, the mixed dispersion was sealed in a 50 mL Teflon-lined autoclave and hydrothermally treated at 200 °C for 20 h. After cooling down to room temperature, the product was centrifuged and washed several times with ethanol and deionized water before drying at 60 °C in an oven. As a control experiment, Fe3O4/G hybrid with different contents of GO (defined as the Fe3O4/G-x, x represent the volume of GO solution) were prepared by varying the volume of GO solution using the same procedure. For comparison, we synthesized bare Fe3O4 microspheres without any GO using the same experimental conditions and a mixture, namely, Fe3O4/G mixed, was prepared by physically mixing Fe3O4 microspheres with graphene powder. To remove the organic ligands, all samples are calcined at 500 °C for 3 h in argon.Materials characterization
X-ray diffraction (XRD) patterns were obtained on a Philips PW1050 X-ray powder diffractometer with Cu Kα radiation (λ = 1.5406 Å). Morphology and microstructure of the products were investigated by transmission electron microscopy (TEM, JEM-1230) at accelerating voltages of 80 kV and scanning electron microscopy (SEM, HITCHI-4800). Structure and selected area electron diffraction were measured by a high-resolution transmission electron microscope (HRTEM, TECNAI G2 F20) operating at 200 kV. Nitrogen adsorption and desorption experiments were carried out using TristarII3020 surface area and porosity analyzer at 77 K. The surface area was calculated using the Brunauer–Emmett–Teller (BET) equation. Pore size distributions were calculated by the Barrett–Joyner–Halenda (BJH) method using the desorption branch of the isotherm curves. Thermogravimetric analysis (TGA) was performed using a CHNS/O analyzer (PE 2400II, Perkin Elmer, America) in air atmosphere from room temperature to 800 °C with a heating rate of 10 °C min−1. The Raman spectra were obtained on DXR SmartRaman (Thermo Fisher, America). All the Fe 2p, O 1s and C 1s XPS spectra were obtained by an Escalab 250Xi spectrometer operating at an Al Kα radiation source.Electrochemical characterization
The electrochemical properties of the obtained F3O4/G, Fe3O4/G mixed and pure Fe3O4 samples were evaluated by assembling CR2032 coin type cells in an argon-filled glove box. The working electrode was prepared by forming slurry of Fe3O4/G, carbon black and sodium alginate in a weight ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:10. Deionized water was used as the solvent. The slurry was coated on copper foil disks and dried overnight in a vacuum oven at 120 °C. Coin cells were fabricated using Li foil as the counter electrode, a porous polyethene film as the separator and LiPF6 (1 M) in ethylene carbonate/dimethyl carbonate/diethyl carbonate (EC/DMC/DEC, 1:1:1 vol%) as the electrolyte. The charge–discharge curves were measured using a Neware Cell tester (Shenzhen Neware Technology Limited Co., China) at a cut off voltage of 0.02–3 V vs. Li/Li+ at different rate at room temperature. The cyclic voltammetry (CV) data were collected with an Arbin electrochemical workstation at a scan rate of 0.1 mV s−1 between 0.005 and 3.0 V. Electrochemical impedance spectroscopy (EIS) measurements were performed on an electrochemical workstation (CHI600E, CH Instruments, Inc.) under a frequency range between 100 kHz and 0.01 Hz with an applied voltage of 5 mV.
:20:10. Deionized water was used as the solvent. The slurry was coated on copper foil disks and dried overnight in a vacuum oven at 120 °C. Coin cells were fabricated using Li foil as the counter electrode, a porous polyethene film as the separator and LiPF6 (1 M) in ethylene carbonate/dimethyl carbonate/diethyl carbonate (EC/DMC/DEC, 1:1:1 vol%) as the electrolyte. The charge–discharge curves were measured using a Neware Cell tester (Shenzhen Neware Technology Limited Co., China) at a cut off voltage of 0.02–3 V vs. Li/Li+ at different rate at room temperature. The cyclic voltammetry (CV) data were collected with an Arbin electrochemical workstation at a scan rate of 0.1 mV s−1 between 0.005 and 3.0 V. Electrochemical impedance spectroscopy (EIS) measurements were performed on an electrochemical workstation (CHI600E, CH Instruments, Inc.) under a frequency range between 100 kHz and 0.01 Hz with an applied voltage of 5 mV.
Results and discussion
The F3O4/G was prepared via a one-pot solvothermal approach based on coordination-driven self-assembly. Fig. 1 shows a plausible model of the 3D framework formation, which can be described as follows. In the first step, Fe-ions could be anchored on graphene sheets through the functional group on the GO surface due to the good miscibility of GO in the ethylene glycol–water suspension. The negatively charged functional groups on the surface of GO could absorb positively charged Fe3+ ions because of electrostatic attraction, which might serve as nucleation and assembly centers for Fe3O4. In fact, every nucleation center could gather a lot of Fe3+ ions and sodium oleate molecules, and more GO would be anchored around the center due to charge balance principle. The second step is a hydrothermal reaction, in which the nucleation and assembly of Fe3O4 occurred simultaneously with the reduction and assembly of GO. In our system, the usage of sodium oleate was very important due to its dual role as a surfactant and reaction assistant. Experiments show that GO will form a hydrogel in ethylene glycol–water systems, which strongly depend on the volume of GO solution (VGO). If VGO was low (e.g., 4 mL), only a black powdery material was produced after hydrothermal reduction (Fig. S1d†). Only when the VGO was increased to 6 mL or above, mechanically stable graphene hydrogels were obtained (Fig. S1b†). However, the threshold of GO assembly decreased dramatically when the assembly of Fe3O4 was introduced into the system and eventually formed a 3D F3O4/G hybrid framework. Even if the added volume of GO was as low as 2 mL, a Fe3O4/G hybrid-gel could still be obtained (Fig. S1e†). This means that the mass loading of active materials in our hybrid product is higher than that of most other Fe3O4–graphene composites in previous reports,33,34 which is of great importance in commercial applications. Moreover, the size of the Fe3O4/G gel was smaller than that of the pure GO gel at the same volume of GO solution (Fig. S1a and b†), which illustrates that the hybrid forms a more dense and stable gel in the presence of the Fe3O4 assembly. In addition, the existence of GO was helpful for forming uniform Fe3O4 hierarchical particles (Fig. S2†). Therefore, we defined this mutual interaction between Fe3O4 and GO as coordination-driven self-assembly. To further confirm this mutual effect, parallel experiments with different Fe sources were carried out. As shown in Fig. S1f,† the hybrid hydrogel could not be produced without the self-assembly of Fe3O4. Moreover, in situ nucleation and/or self-assembly can lead to a strong binding force between Fe3O4 and GO, which is beneficial to electronic transmission and structural integrity. | ||
| Fig. 1 Schematic illustration of the synergistic effect of self-assembly and the formation of Fe3O4/G hybrid 3D framework. | ||
The crystallographic structure and phase purity of the F3O4/G, Fe3O4/G mixed and bare Fe3O4 were determined by X-ray powder diffraction (XRD), as shown in Fig. 2. It was found that all the diffraction peaks can be well indexed to the magnetic cubic structure of Fe3O4 (JCPDS 79-0419). The mean crystallite size of the Fe3O4/G and Fe3O4/G mixed are 18.6 and 23.6 nm, respectively, as calculated using the Scherrer equation according to the (311) peaks. In addition, a diffraction peak at ∼26° corresponding to graphene was observed in the pattern of Fe3O4/G mixed, which can be ascribed to the reduction and stacking of GO. Remarkably, no apparent peak can be identified at 20–30° in the Fe3O4/G sample, indicating that the Fe3O4 spheres are efficiently deposited on the surface of graphene, suppressing the stacking of graphene layers, which reflects the fact that the F3O4/G is composed of few-layer stack graphene sheets.
 | ||
| Fig. 2 XRD patterns of the Fe3O4/G, Fe3O4/G mixed and bare Fe3O4. | ||
The morphologies and microstructure of the samples were characterized by SEM and TEM. As shown in Fig. 3a and b, without the presence of GO, the bare Fe3O4 spheres are constructed by primary nanoparticles and have a wide size distribution of 100–500 nm. Fig. 3c is a typical SEM image of the Fe3O4/G framework. From the image it is clear that the Fe3O4 spheres are uniformly wrapped by graphene sheets and each of them has a diameter of about 210 nm (Fig. S2†). The TEM image also reveals that these Fe3O4 particles are firmly attached to the graphene sheet, even after extended sonication was used to disperse the Fe3O4/G hybrid framework for TEM characterization. We believe that this perfect combination prevents the agglomeration of nanoparticles and also enables fast electron transport through the underlying graphene layers to Fe3O4, guaranteeing the efficient electrochemical performance. The high-resolution TEM images in Fig. 3e and f show the interfacial structure between the graphene and Fe3O4 spheres. The well-resolved lattice fringes with an interplane distance of 0.253 nm are caused by the (311) plane of Fe3O4. It should be emphasized that the volume of GO solution is a critical factor in our one-pot synthesis approach. We found that when the volume of the GO solution was 6 mL, the Fe3O4 spheres would be anchored by graphene sheets to form hybrid hydrogels and do not present an obstacle to the self-assembly of Fe3O4 (Fig. 2c and d and S3b†). A small amount of GO solution would result in ineffective wrapping around the Fe3O4 spheres (Fig. S3a†), while a large amount of GO solution would damage the assembled structure of the Fe3O4 spheres (Fig. S3c and 3d†). Therefore, we chose the Fe3O4/G (6 mL GO solution) sample for the following structure and properties characterization.
 | ||
| Fig. 3 (a) and (b) are the SEM and TEM images of bare Fe3O4 spheres; (c) and (d) are the SEM and TEM images of Fe3O4/G framework; (e) is a magnified image of a single Fe3O4/G composite, and (f) is a HRTEM image of Fe3O4/G framework. | ||
Fig. 4a presents N2 adsorption–desorption isotherms for Fe3O4/G at 77 K. The isotherm can be identified as a type-IV isotherm with an H3 hysteresis loop that is attributed to a mesoporous structure, which indicates that the Fe3O4 spheres have hierarchical structure. The pore size distribution for Fe3O4/G is shown in the inset of Fig. 4a and it demonstrates the formation of randomly distributed pores with sizes from 4 to 16 nm with dominant 4 and 8 nm pores. The isotherms of Fe3O4/G mixed and pure Fe3O4 have similar results, as shown in Fig. S4a–d.† Although the BET specific area of Fe3O4/G is almost the same for both Fe3O4/G mixed and pure Fe3O4, the total pore volume of Fe3O4/G is about two times larger than that of the two others as shown in Fig. 4b. Combined with the TEM and SEM images, we conclude the pore structures may benefit electrolyte ion diffusion to active sites with less resistance and tolerate the volume changes of Fe3O4/G during the charge–discharge cycles.
 | ||
| Fig. 4 (a) N2 adsorption–desorption isotherms of the Fe3O4/G sample; (b) comparison of the BET surface areas and total pore volumes of the Fe3O4/G, Fe3O4/G mixed and bare Fe3O4 samples. The inset in (a) shows the pore size distribution calculated by the BJH method. | ||
X-ray photoelectron spectroscopy (XPS) analysis was employed to further characterize the product. The wide scan XPS spectrum of the Fe3O4/G hybrid framework demonstrates the presence of Fe, O, and C elements (Fig. S5a†). Fig. 5a–b exhibits the high-resolution XPS spectrum of Fe3O4/G. In Fig. 5a, the Fe 2p1/2 and 2p3/2 peaks located at around 711 and 724.9 eV are observed, which indicates that the presence of both the Fe2+ and Fe3+ ions in Fe3O4. For carbon, the C 1s XPS profiles in Fig. 2c not only shows the presence of the carbon element but also reveals the efficient reduction of GO. After solvothermal reaction, the sp2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds at 284.5 eV increased, while the epoxy C–O bonds at 286.1 eV and CO bonds at 288.3 eV decreased, indicating an efficient reduction.35 The structural and electronic properties of graphene in the Fe3O4/G hybrid have been investigated using laser Raman spectroscopy. For comparison, we also measured the Fe3O4/G mixed and bare Fe3O4 and the results are shown in Fig. 5c. Both the Fe3O4/G hybrid and Fe3O4/G mixed show two prominent peaks, one is the G band at about 1600 cm−1, which corresponds to ordered sp2 bond carbon, and the other is the D band at around 1350 cm−1, which results from edges, other defects and disordered carbon in the symmetrical hexagonal graphitic lattice.36 Compared to the Fe3O4/G mixed sample, the shift of the peaks can be found for both D and G bands in Fe3O4/G hybrid, indicating a significant charge transfer between the graphene nanosheets and Fe3O4 spheres.15 The appearance of weak D and G peaks of the bare Fe3O4 can be ascribed to the carbonization of ligands during calcining. The content of Fe3O4 in the composite was evaluated by thermogravimetric (TG) analysis, with the results shown in Fig. 5d. The weight loss of ∼0.4% up to 150 °C is attributed to the release of water adsorbed on the samples. The weight loss that occurs during the heat treatment from room temperature to 800 °C is attributed to the oxidation of graphene/carbon, and a weight gain afterwards is believed to correspond to the oxidation of Fe3O4 in air. As for bare Fe3O4, no obvious weight loss is found, indicating that the weight gain (oxidation of Fe3O4) is equal to the weight loss (oxidation of amorphous). Therefore, the amorphous carbon in bare Fe3O4 can be derived to be about 4 wt%. For the Fe3O4/G and Fe3O4/G mixed samples, based on the total weight loss of carbon materials (9.6% and 8.4%), the original weight fraction of Fe3O4 is calculated as 86.4% and 87.6%, respectively, and the content of graphene in Fe3O4/G and Fe3O4/G mixed are calculated to be 5.6% and 4.4%, which is approximately consistent with the theoretical calculations. TG results show that the Fe3O4/G hybrid framework has a relatively high mass loading of active materials compared with other Fe3O4–graphene composites (Table 1).
C bonds at 284.5 eV increased, while the epoxy C–O bonds at 286.1 eV and CO bonds at 288.3 eV decreased, indicating an efficient reduction.35 The structural and electronic properties of graphene in the Fe3O4/G hybrid have been investigated using laser Raman spectroscopy. For comparison, we also measured the Fe3O4/G mixed and bare Fe3O4 and the results are shown in Fig. 5c. Both the Fe3O4/G hybrid and Fe3O4/G mixed show two prominent peaks, one is the G band at about 1600 cm−1, which corresponds to ordered sp2 bond carbon, and the other is the D band at around 1350 cm−1, which results from edges, other defects and disordered carbon in the symmetrical hexagonal graphitic lattice.36 Compared to the Fe3O4/G mixed sample, the shift of the peaks can be found for both D and G bands in Fe3O4/G hybrid, indicating a significant charge transfer between the graphene nanosheets and Fe3O4 spheres.15 The appearance of weak D and G peaks of the bare Fe3O4 can be ascribed to the carbonization of ligands during calcining. The content of Fe3O4 in the composite was evaluated by thermogravimetric (TG) analysis, with the results shown in Fig. 5d. The weight loss of ∼0.4% up to 150 °C is attributed to the release of water adsorbed on the samples. The weight loss that occurs during the heat treatment from room temperature to 800 °C is attributed to the oxidation of graphene/carbon, and a weight gain afterwards is believed to correspond to the oxidation of Fe3O4 in air. As for bare Fe3O4, no obvious weight loss is found, indicating that the weight gain (oxidation of Fe3O4) is equal to the weight loss (oxidation of amorphous). Therefore, the amorphous carbon in bare Fe3O4 can be derived to be about 4 wt%. For the Fe3O4/G and Fe3O4/G mixed samples, based on the total weight loss of carbon materials (9.6% and 8.4%), the original weight fraction of Fe3O4 is calculated as 86.4% and 87.6%, respectively, and the content of graphene in Fe3O4/G and Fe3O4/G mixed are calculated to be 5.6% and 4.4%, which is approximately consistent with the theoretical calculations. TG results show that the Fe3O4/G hybrid framework has a relatively high mass loading of active materials compared with other Fe3O4–graphene composites (Table 1).
 | ||
| Fig. 5 XPS spectra of core-level Fe 2p (a) and C 1s (b) for Fe3O4/G hybrid framework; Raman spectra (c) and TGA curves (d) of Fe3O4/G, Fe3O4/G mixed and bare Fe3O4 samples. | ||
| Materials | Ratio | Cycles (density) | Capacity | Retention | Ref. |
|---|---|---|---|---|---|
| a (1 C = 1 A g−1). Ratio: the weight percentage of active material in composites. Retention: the capacity retention from the second to the end of cycles. | |||||
| Fe3O4–graphene | 87.6% | 500 (0.5 C) | 1164 | 99.4% | This work |
| 50 (1 C) | 831 | — | |||
| Fe3O4–graphene | 67% | 300 (5.2 C) | 577 | ∼96.1% | 34 |
| Fe3O4–graphene | 70% | 50 (1 C) | 612 | ∼47.1% | 32 |
| Fe3O4–graphene | 40% | 50 (0.05 C) | 675 | ∼73.4% | 31 |
| Fe3O4–graphene | 87.2% | 300 (0.1 C) | 1107 | ∼83.5% | 28 |
| Fe3O4–C–graphene | 50.2% | 100 (0.1 C) | 660 | ∼72.1% | 54 |
| Fe3O4–graphene | 87.7% | 100 (0.7 C) | ∼560 | ∼91.0% | 30 |
| Fe3O4–graphene | ∼70% | 30 (0.2 C) | 1100 | ∼91.6% | 12 |
| Fe3O4–C | 45.4% | 100 (0.05 C) | 610 | ∼55.5% | 38 |
| Fe3O4–C | 83% | 100 (1 C) | 831 | ∼83.1% | 41 |
| Fe3O4–C | 89.8% | 120 (0.2 C) | 1232 | 107% | 40 |
In order to characterize the electrochemical properties of the samples, half-cells with the lithium plate as the counter electrode were fabricated. Cyclic voltammograms (CVs) of the Fe3O4/G and bare Fe3O4 were measured between 0.005 and 3 V at a scan rate of 0.1 mV s−1. As seen in Fig. 6a and Fig. S6a,† it is obvious that both the samples demonstrate similar well defined redox peaks signifying the lithiation and delithiation of Fe3O4. In the first scan, the cathodic peak appearing at 0.52 V is attributed to the reduction of Fe3+ and Fe2+ to Fe0 (Fe3O4 + 8Li → 3Fe + 4Li2O) and the formation of SEI layer. In the first anodic process, two peaks were observed at 1.65 and 1.85 V in Fe3O4/G, attributed to the oxidation of Fe0 to Fe2+ and Fe3+, respectively.37,38 It is clear that the subsequent cycles are quite different from the first one. Both the anodic and cathodic peaks are positively shifted, which is attributed to structural modification after the first cycle due to lithium insertion and extraction.39 It can be noted that the cathodic peaks at 0.75 V and the anodic peaks located at about 1.70 and 1.96 V for the second or fifth cycle of the Fe3O4/G electrode almost overlap, indicating the high reversibility of electrode.40,41 The charge–discharge curves of the Fe3O4/G hybrid and bare Fe3O4 electrodes for the 1st, 2nd, 100th, 200th, and 500th cycles are illustrated in Fig. 6b and Fig. S6b.† As shown in Fig. 6b, the first discharge and charge capacities of Fe3O4/G are measured to be 1573 and 1113 mA h g−1, respectively, resulting in an initial Coulombic efficiency of 72.4%. The bare Fe3O4 exhibits a similar electrochemical performance at the first cycle, with the first discharge and charge capacities of 1216 and 924 mA h g−1, producing a Coulombic efficiency of 75.9% (Fig. S6b†). We note that the first discharge capacity is much higher than that of the theoretical value for Fe3O4 and graphene; moreover, the reasons for exceeding theoretical capacities can be ascribed to the formation of a solid electrolyte interphase resulting from electrolyte degradation at low voltage and additional interfacial/active site Li storage mechanism due to nano-scale and mesoporous structures, as well as the synergistic effect between the graphene and the Fe3O4 particles.42,43 The initial Coulombic efficiency of bare Fe3O4 is slightly higher than that of Fe3O4/G, which may be ascribed to the lower surface area of bare Fe3O4 (Fig. 4b). The irreversible capacity loss can be attributed to the inevitable formation of a surface SEI layer accompanying the electrolyte decomposition during discharge. The long plateau at 0.7 V in the initial discharge curve corresponds to the reversible reduction from Fe3+ (or Fe2+) to Fe0 [LixFe3O4 + (8 − x)Li+ + (8 − x)e− → 4Li2O + 3Fe] and the irreversible formation of a solid electrolyte interphase film. From the second cycle, this plateau increases to a slightly higher voltage and becomes much shorter, indicating that different lithiation reactions take place between the first and the following cycles and the existence of irreversible reactions. With cycling, no obvious changes to either charge or discharge profiles are observed, indicating a highly reversible electrode reaction process and a stable electrode structure characteristic. In the case of the bare Fe3O4, although the first discharge curve is very similar to that of the Fe3O4/G, the specific capacity of the bare Fe3O4 electrode decreases rapidly due to volume expansion in the subsequent charge–discharge cycles, as shown in Fig. S6b.†
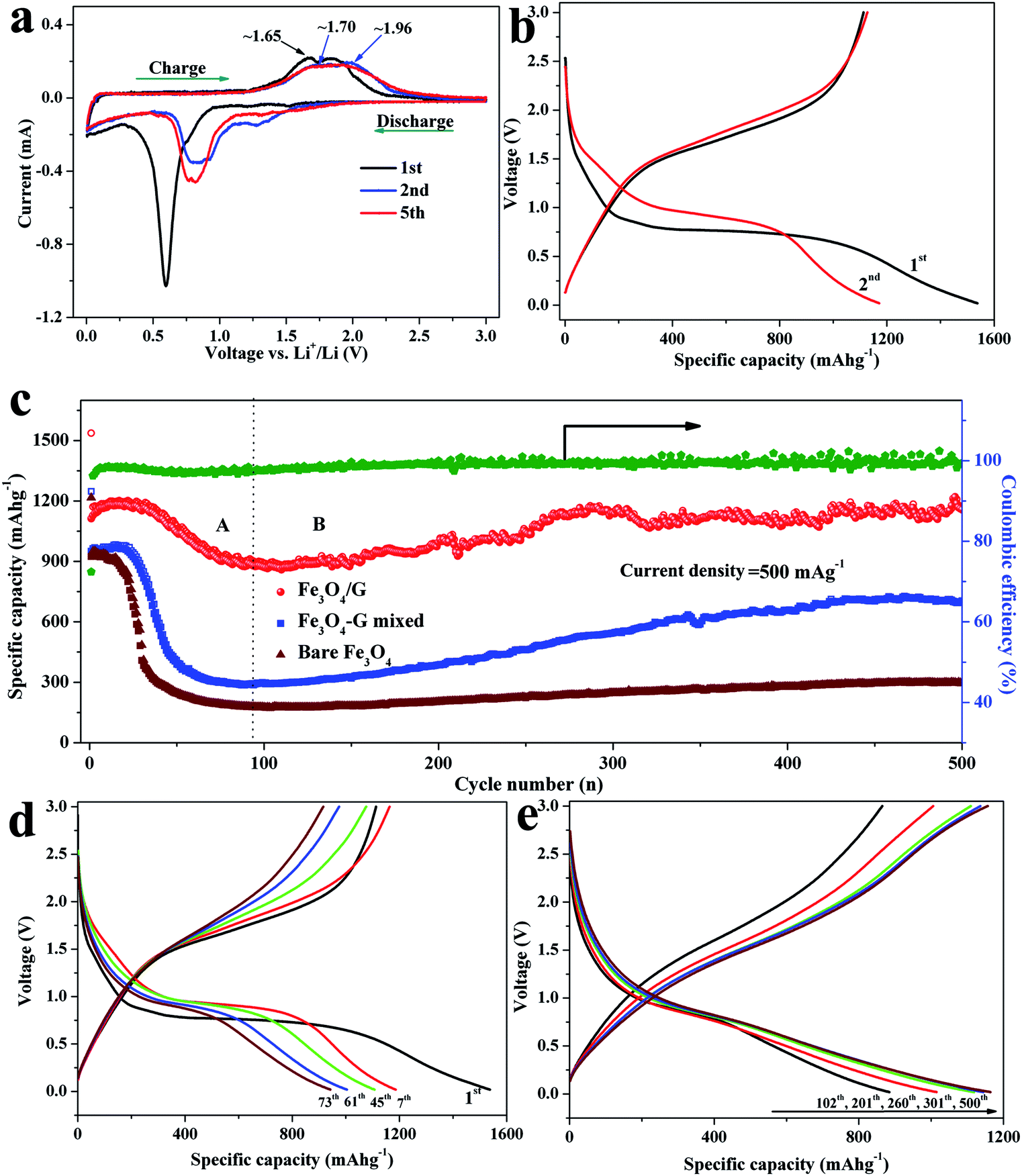 | ||
| Fig. 6 (a) Cyclic voltammogram curves of Fe3O4/G hybrid at a scan rate of 0.1 mV s−1; (b) charge and discharge profiles of the Fe3O4/G hybrid at a current density of 500 mA g−1 between 0.02 and 3 V; (c) cycling performance and Coulombic efficiency of Fe3O4/G, Fe3O4/G mixed and bare Fe3O4 samples. Charge–discharge curves of Fe3O4/G anode for: (d) stage A; (e) stage B. | ||
Fig. 6c shows the long-term cycling performance and the corresponding Coulombic efficiency of the Fe3O4/G hybrid framework and bare Fe3O4 electrodes in a potential window of 0.02–3 V vs. Li+/Li at a current density of 500 mA g−1. In order to clearly demonstrate the superior electrochemical performance and synergistic effect in Fe3O4/G, we prepared the Fe3O4/G mixed sample by physically mixing graphene with Fe3O4 spheres to offer comparisons with the Fe3O4/G electrode. In our study, the capacity is calculated based on the total mass of the composite, including Fe3O4 and graphene sheets. As can be seen, the Fe3O4/G electrode exhibits higher capacity and good cyclic retention compared to Fe3O4/G mixed and bare Fe3O4. The initial discharge capacities are 1537, 1247 and 1216 mA h g−1 for Fe3O4/G, Fe3O4/G mixed and bare Fe3O4, respectively. After 500 cycles, the Fe3O4/G still delivers a discharge of 1164 mA h g−1, while the capacities of Fe3O4/G mixed and bare Fe3O4 decrease dramatically to 702 and 297 mA h g−1, respectively. Moreover, Fe3O4/G still possesses better lithium storage properties even compared with Fe3O4/G mixed (the same graphene content), indicating that there exists a strong interaction force between Fe3O4 and graphene, which is consistent with the TEM and Raman results. In addition, the Coulombic efficiency of Fe3O4/G increased rapidly after several cycles and steadily reached up to 99%. Moreover, it can be noted that the specific capacity undergoes a gradual rising process after the slow fading process in the first 80 cycles. Such a phenomenon of increasing capacity upon cycling has been found in Fe3O4 and other metal oxide anode materials.29,44–48 To clarify the mechanism, the cycle curve of Fe3O4/G hybrid was divided into two different steps based on the variation of the specific capacity, as illustrated in Fig. 6c–e. Stage (A): an obvious drop in specific capacity was recorded, which may be ascribed to the cracking of some of un-anchored Fe3O4 particles. Stage (B): the specific capacity stabilizes in the range of 80–150 cycles and increases after 170 cycles. This may be caused by three factors as follows. First, several discharge–charge cycles are needed to form a stable SEI film, allowing the greigite crystals to percolate throughout and establish intimate contacts with the current collector.49,50 Second, the slight increase of capacity is due to the reversible growth of a polymeric gel-like film caused by the decomposition of electrolytes at a low potential, which enables mechanical cohesion and delivers excess capacity through a so-called “pseudo-capacity-type” mechanism. Last but not least, an anomalous monotonic increase in the discharge capacity was observed from 884 (102th) and 1015 (201th) to 1118 mA h g−1 (260th) and is followed by a nearly constant capacity at 1160 mA h g−1 till the 500th cycle. The voltage profiles are shown in Fig. 6e. The discharge curves overlap basically at ∼0.8 V, which indicates the sustained excellent reversibility of the traditional conversion mechanism in Fe3O4/G anode. However, the curve's decay below 0.8 V slows down and the voltage hysteresis becomes narrower as confirmed by the closer discharge–charge voltage profiles with cycling, which results from faster kinetics and an improved energy efficiency.51,52 The electrochemical properties of the Fe3O4/G hybrid framework prepared in the present study and those of the iron oxide materials reported in the literature are summarized in Table 1. It can be seen that the Fe3O4/G hybrid electrode is of high capacity and excellent cycling stability, as well as a high ratio of active materials in composites. The reasons for high specific capacity and good cycling stability of the Fe3O4/G hybrid can be ascribed to the following factors: (1) the hierarchical structure and uniform size distribution of Fe3O4 spheres can alleviate stress due to volume expansion in the discharge–charge process, (2) the incorporation of graphene not only enhances the conductivity of electrode, but also partially accommodates the volume change during cycling, (3) the strong adhesive force of Fe3O4 to graphene empowers fast electron transport through graphene to enhance the electrochemical performance. (4) The 3D hybrid framework constructed by hierarchical Fe3O4 spheres and flexible graphene sheets possesses a large number of pores and large electrolyte/electrode interface area, which suggests that it has more channels for Li-ion diffusion and reversible lithium storage site and (5) the Fe3O4 crystals are uniformly anchored among the network of graphene, which inhibits pulverization and maintains the integrity of electrode.
Benefiting from the unique structure of Fe3O4/G hybrid framework, the Fe3O4/G electrode exhibits exceptional rate performance. As shown in Fig. 7a, the electrode delivers capacities of 1280, 1240, 1145, 956, 831 and 510 mA h g−1 when cycled at high rate of 100, 200, 400, 800, 1000 and 2000 mA g−1, respectively, which present a superior rate performance in comparison with presently reported results. When the charge–discharge system is back to the 200 mA g−1, the discharge capacities can recover. To demonstrate the synergistic effect between Fe3O4 and graphene, the rate capability of Fe3O4/G mixed is also investigated under the same condition. Compared to the Fe3O4/G electrode, Fe3O4/G mixed electrode delivers much lower specific capacities 200–1000 mA h g−1 at current rates of 100–2000 mA g−1 as a result of inefficient electron and lithium-ion transportation between Fe3O4 and graphene. As for the bare Fe3O4 electrode, the reversible capacity continuously decayed with cycling, and exhibited a capacitance less than 50 mA h g−1 at a current of 2000 mA g−1. In addition, the Fe3O4/G hybrid exhibits excellent rate cyclic performance, because there is no significant capacity loss at each rate test.
 | ||
| Fig. 7 (a) Rate performance of Fe3O4/G, Fe3O4/G mixed and bare Fe3O4 cycled between 0.02 and 3 V. (b) Nyquist plots at the OCP (5 mV perturbation) for the electrodes. | ||
As an important index for insight into the electrochemical behaviour, electrochemical impedance spectroscopy (EIS) was conducted to identify the relationship between the electrochemical performance and electrode kinetics. All the coin cells are cycled by rate testing before EIS testing. The Nyquist plots for the samples are shown in Fig. 7b with a frequency range from 100 kHz to 0.01 Hz, which share the common feature of a depressed semicircle at high frequency and an inclined line at low frequency. The diameter of the semicircle for the Fe3O4/G hybrid framework electrode in the high-medium frequency region is much smaller than that of Fe3O4/G mixed or bare Fe3O4, indicating that the former electrodes possess the minimum charge transfer resistance (Rct) among the three samples. This means that the rapid electron transfer during the electrochemical reaction of the composite can be ascribed to the increased contact area and strong binding force between Fe3O4 and graphene as well as the enhanced electrical conductivity of the overall electrode.53,54 In the low frequency region, a more vertical straight line of Fe3O4/G compared to Fe3O4/G mixed and bare Fe3O4 is further evidence for the faster Li+ ion diffusion behavior of the Fe3O4/G hybrid framework electrodes. The Fe3O4/G hybrid framework presents satisfying electrochemical kinetics properties, leading to a remarkable improvement in cyclic stability and rate properties.
As shown by the results presented above, the Fe3O4/G hybrid electrode displays excellent electrochemical performance and structural stability. These outstanding properties can be attributed to several potential factors. Firstly, the 3D Fe3O4/G hybrid frameworks provide a large surface area and efficiently reduce the diffusion length of both electrons and lithium ions. Secondly, graphene sheets provide a highly conductive matrix for electron transfer during the lithiation and delithiation processes. Moreover, the intimate interaction between Fe3O4 and graphene sheets prevents the aggregation of Fe3O4 nanoparticles and the restacking of graphene nanosheets.
Conclusion
In summary, we have demonstrated the coordination-driven self-assembly of Fe3O4 with graphene sheets under hydrothermal conditions for simple in situ synthesis of a 3D Fe3O4/G hybrid architecture. The key factors of our approach are the introduction of ethylene glycol–water system and the addition of sodium oleate, which can ensure the occurrence of self-assembly of both Fe3O4 and graphene oxide. The two assembly processes mutually reinforced and interacted with each other, obtaining the integral Fe3O4/G hybrid framework. The Fe3O4/G hydrogel could be formed at a very low concentration of graphene oxide due to the coordination-driven self-assembly, which indicates the high mass proportion of active materials in a composite. Such a special and unique architecture established by the inter-dispersion of Fe3O4 spheres provides better protection against aggregation and volume changes of the active material, guarantees more lithium-storage sites, and ensures favourable transport kinetics for both electrons and lithium ions. As a result, the Fe3O4/G hybrid frameworks demonstrate outstanding enhancement of durability and rate performance compared with Fe3O4/G mixed and bare Fe3O4, with a very high reversible capacity of 1164 mA h g−1 even after 500 cycles at a current density of 500 mA g−1 and 510 mA h g−1 at a high rate of 2000 mA g−1. It is believed that this approach can be readily applied to other metal oxide–GO hybrid nanostructures in order to study their synergetic and self-assembly properties.Acknowledgements
The authors gratefully acknowledge the financial support for this study from the National Natural Science Foundation of China (no. 51272231), and the Doctoral Fund of the Ministry of Education of China (no. 20100101110039).Notes and references
- K. Kang, Y. S. Meng, J. Bréger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef CAS PubMed.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- M. Wu, J. E. Sabisch, X. Song, A. M. Minor, V. S. Battaglia and G. Liu, Nano Lett., 2013, 13, 5397–5402 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J.-M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- L. Zhang, H. B. Wu, Y. Yan, X. Wang and X. W. Lou, Energy Environ. Sci., 2014, 7, 3302–3306 CAS.
- C. Zhu, D. Chao, J. Sun, I. M. Bacho, Z. Fan, C. F. Ng, X. Xia, H. Huang, H. Zhang, Z. X. Shen, G. Ding and H. J. Fan, Adv. Mater. Interfaces, 2015, 2, 1400499 Search PubMed.
- W. Wen, J.-M. Wu and M.-H. Cao, Nanoscale, 2014, 6, 12476–12481 RSC.
- L. Li, H. B. Wu, L. Yu, S. Madhavi and X. W. D. Lou, Adv. Mater. Interfaces, 2014, 1, 1400050 Search PubMed.
- X. Li, A. Dhanabalan and C. Wang, J. Power Sources, 2011, 196, 9625–9630 CrossRef CAS PubMed.
- H. Wang, Z. Xu, H. Yi, H. Wei, Z. Guo and X. Wang, Nano Energy, 2014, 7, 86–96 CrossRef CAS PubMed.
- W. Chen, S. Li, C. Chen and L. Yan, Adv. Mater., 2011, 23, 5679–5683 CrossRef CAS PubMed.
- B. Lim, J. Jin, J. Yoo, S. Y. Han, K. Kim, S. Kang, N. Park, S. M. Lee, H. J. Kim and S. U. Son, Chem. Commun., 2014, 50, 7723–7726 RSC.
- S. H. Choi, Y. N. Ko, K. Y. Jung and Y. C. Kang, Chemistry, 2014, 20, 11078–11083 CrossRef CAS PubMed.
- C. Ban, Z. Wu, D. T. Gillaspie, L. Chen, Y. Yan, J. L. Blackburn and A. C. Dillon, Adv. Mater., 2010, 22, E145–E149 CrossRef CAS PubMed.
- Y. Piao, H. S. Kim, Y. E. Sung and T. Hyeon, Chem. Commun., 2010, 46, 118–120 RSC.
- S. Jin, H. Deng, D. Long, X. Liu, L. Zhan, X. Liang, W. Qiao and L. Ling, J. Power Sources, 2011, 196, 3887–3893 CrossRef CAS PubMed.
- Q. Q. Xiong, J. P. Tu, Y. Lu, J. Chen, Y. X. Yu, Y. Q. Qiao, X. L. Wang and C. D. Gu, J. Phys. Chem. C, 2012, 116, 6495–6502 CAS.
- C. Yuan, H. B. Wu, Y. Xie and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 1488–1504 CrossRef CAS PubMed.
- S. H. Lee, S. H. Yu, J. E. Lee, A. Jin, D. J. Lee, N. Lee, H. Jo, K. Shin, T. Y. Ahn, Y. W. Kim, H. Choe, Y. E. Sung and T. Hyeon, Nano Lett., 2013, 13, 4249–4256 CrossRef CAS PubMed.
- Y. Li, Z.-Y. Fu and B.-L. Su, Adv. Funct. Mater., 2012, 22, 4634–4667 CrossRef CAS PubMed.
- X. Fan, J. Shao, X. Xiao, L. Chen, X. Wang and S. Li, J. Mater. Chem. A, 2014, 2, 14641–14648 CAS.
- Q. Liu, Z. F. Li, Y. Liu, H. Zhang, Y. Ren, C. J. Sun, W. Lu, Y. Zhou, L. Stanciu, E. A. Stach and J. Xie, Nat. Commun., 2015, 6, 6127 CrossRef PubMed.
- F. Wu, R. Huang, D. Mu, B. Wu and S. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 19254–19264 CAS.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2014, 14, 271–279 CrossRef PubMed.
- J. Yao, X. Shen, B. Wang, H. Liu and G. Wang, Electrochem. Commun., 2009, 11, 1849–1852 CrossRef CAS PubMed.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- J. Yin, H. Shi, P. Wu, Q. Zhu, H. Wang, Y. Tang, Y. Zhou and T. Lu, New J. Chem., 2014, 38, 4036–4040 RSC.
- Q. Zhou, Z. Zhao, Z. Wang, Y. Dong, X. Wang, Y. Gogotsiac and J. Qiu, Nanoscale, 2014, 6, 2286–2291 RSC.
- G. Zhou, D.-W. Wang, F. Li, L. Zhang, N. Li, Z.-S. Wu, L. Wen, G. Q. Lu and H.-M. Cheng, Chem. Mater., 2010, 22, 5306–5313 CrossRef CAS.
- M. Sathish, T. Tomai and I. Honma, J. Power Sources, 2012, 217, 85–91 CrossRef CAS PubMed.
- S. Bhuvaneswari, P. M. Pratheeksha, S. Anandan, D. Rangappa, R. Gopalan and T. N. Rao, Phys. Chem. Chem. Phys., 2014, 16, 5284–5294 RSC.
- T.-q. Wang, X.-l. Wang, Y. Lu, Q.-q. Xiong, X.-y. Zhao, J.-b. Cai, S. Huang, C.-d. Gu and J.-p. Tu, RSC Adv., 2014, 4, 322–330 RSC.
- L. Fan, B. Li, D. W. Rooney, N. Zhang and K. Sun, Chem. Commun., 2015, 51, 1597–1600 RSC.
- H.-K. Kim, S.-H. Park, S.-B. Yoon, C.-W. Lee, J. H. Jeong, K. C. Roh and K.-B. Kim, Chem. Mater., 2014, 26, 4838–4843 CrossRef CAS.
- R. Chen, T. Zhao, W. Wu, F. Wu, L. Li, J. Qian, R. Xu, H. Wu, H. M. Albishri, A. S. Al-Bogami, D. A. El-Hady, J. Lu and K. Amine, Nano Lett., 2014, 14, 5899–5904 CrossRef CAS PubMed.
- H.-S. Lim, B.-Y. Jung, Y.-K. Sun and K.-D. Suh, Electrochim. Acta, 2012, 75, 123–130 CrossRef CAS PubMed.
- G. Chen, M. Zhou, J. Catanach, T. Liaw, L. Fei, S. Deng and H. Luo, Nano Energy, 2014, 8, 126–132 CrossRef CAS PubMed.
- M. M. Thackeray, J. Am. Ceram. Soc., 1999, 82, 3347–3354 CrossRef CAS PubMed.
- G. Gao, S. Lu, B. Dong, Z. Zhang, Y. Zheng and S. Ding, J. Mater. Chem. A, 2015, 3, 4716–4721 CAS.
- L. Shen, H. Song, H. Cui, X. Wen, X. Wei and C. Wang, CrystEngComm, 2013, 15, 9849–9854 RSC.
- S. Xu, C. M. Hessel, H. Ren, R. Yu, Q. Jin, M. Yang, H. Zhao and D. Wang, Energy Environ. Sci., 2014, 7, 632 CAS.
- J. Morales, L. Sánchez, F. Martín, F. Berry and X. Ren, J. Electrochem. Soc., 2005, 152, A1748 CrossRef CAS PubMed.
- X. Li, L. Qiao, D. Li, X. Wang, W. Xie and D. He, J. Mater. Chem. A, 2013, 1, 6400–6406 CAS.
- P. L. Taberna, S. Mitra, P. Poizot, P. Simon and J. M. Tarascon, Nat. Mater., 2006, 5, 567–573 CrossRef CAS PubMed.
- J.-S. Do and C.-H. Weng, J. Power Sources, 2005, 146, 482–486 CrossRef CAS PubMed.
- Y. Xu, J. Guo and C. Wang, J. Mater. Chem., 2012, 22, 9562–9567 RSC.
- Y.-Y. Hu, Z. Liu, K.-W. Nam, O. J. Borkiewicz, J. Cheng, X. Hua, M. T. Dunstan, X. Yu, K. M. Wiaderek, L.-S. Du, K. W. Chapman, P. J. Chupas, X.-Q. Yang and C. P. Grey, Nat. Mater., 2013, 12, 1130–1136 CrossRef CAS PubMed.
- G. Li, B. Zhang, F. Yu, A. A. Novakova, M. S. Krivenkov, T. Y. Kiseleva, L. Chang, J. Rao, A. O. Polyakov, G. R. Blake, R. A. de Groot and T. T. M. Palstra, Chem. Mater., 2014, 26, 5821–5829 CrossRef CAS.
- Y. Yu, C.-H. Chen, J.-L. Shui and S. Xie, Angew. Chem., Int. Ed., 2005, 44, 7085–7089 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- F. Ma, A. Yuan and J. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 18129–18138 CAS.
- B. G. Choi, M. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020–4028 CrossRef CAS PubMed.
- S. Tao, W. Yue, M. Zhong, Z. Chen and Y. Ren, ACS Appl. Mater. Interfaces, 2014, 6, 6332–6339 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra04837k |
| This journal is © The Royal Society of Chemistry 2015 |