
High-performance recyclable V–N–C catalysts for the direct hydroxylation of benzene to phenol using molecular oxygen†
Sensen. Shangab,
Bo Chenab,
Lianyue Wanga,
Wen Daia,
Yi Zhanga and
Shuang Gao*a
aDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian National Laboratory for Clean Energy, DNL, Dalian, 116023, People's Republic of China. E-mail: sgao@dicp.ac.cn; Fax: +86-0411-84379728; Tel: +86-0411-84379728
bUniversity of Chinese Academy of Sciences, Beijing, 10049, People's Republic of China
First published on 30th March 2015
Abstract
In this report, high-performance recyclable V–N–C catalysts for the direct hydroxylation of benzene to phenol using molecular oxygen were designed and prepared. Up to 12.6% yield of phenol with selectivity as high as 97.8% was achieved employing V–N–C-600 catalyst in acetonitrile. The catalytic recycling tests demonstrated that the V–N–C-600 catalyst exhibited high potential for reusability. The catalysts were characterized systematically by thermogravimetric analysis, Fourier transform infrared spectroscopy, N2 adsorption–desorption, powder X-ray diffraction, Raman spectroscopy, X-ray photoelectron spectroscopy techniques and scanning electron microscopy. It was determined that the interaction between surface nitrogen of the supports and reactive vanadium species played an important role in the excellent stability in recycling of the heterogeneous V–N–C catalysts.
1. Introduction
Phenol is a crucial bulk commodity chemical in industry, which is widely used in the manufacturing of phenol resins, caprolactam, adipic acid, fibers, and many other chemicals.1–3 Nowadays, more than 90% of phenol is still produced industrially by the three-step cumene process,4 which involves unexpected high energy consumption and significant formation of side products such as acetone and methylstyrene. Therefore, the direct hydroxylation of benzene to phenol is potentially an area of great interest and is still regarded as one of the 10 most difficult challenges in catalysis,5 as the phenol selectivity is limited due to the higher oxidation reactivity of phenol when compared to benzene.6 Indeed, endeavors have been made using green oxidants, such as O2,7–10 H2O2 (ref. 11–18) or N2O.19–23 As for N2O, despite its high selectivity to phenol, the main restriction for the wide application of N2O is its complex operation process.17 On the other hand, H2O2 is too expensive to be applied in commercial production. In this regard, it is highly desirable to develop an efficient catalytic system for the direct hydroxylation of benzene to phenol using O2 as green and economical oxidant agent.24In the past several decades, various catalytic systems, such as membrane process, high-temperature gas-phase and low-temperature liquid-phase catalytic reactions have been developed for this aerobic oxidation. After Mizukami et al. reported the direct hydroxylation of benzene to phenol in gas phase with molecular oxygen activated by dissociated hydrogen obtained from a palladium membrane at 2002,24 Zhang et al. have been devoted to overcoming the instability of Pd membrane.25,26 Meanwhile, the utilization of noble metals largely increased the cost of the catalytic system. In addition, both the vast energy consumption and the unexpected hazard were caused by high-temperature gas-phase catalytic system. Consequently, developing low-temperature liquid-phase oxidation of benzene to phenol catalyzed by highly efficient non-noble metal catalysts, mainly containing Cu or V species, is drawing more and more attention from an economic and environmental point of view. Ohtani et al. tested a series of zeolites loaded with Cu as catalysts with molecular oxygen under atmospheric pressure at room temperature, giving a phenol yield of 1.69% with Cu–NaY as the best catalyst and ascorbic acid as a reducing agent at 1995.27 After then, the Cu ion-exchanged HPMCM-41 catalysts were proved to be more active than the corresponding Cu catalysts supported on SiO2, TiO2, MgO, or NaY, even though with less than the phenol yield of 2%.28 It was worth mentioning that the accumulation of H2O2 was confirmed during the liquid-phase oxidation process. Moreover, the yield of phenol over CuO–Al2O3 and Cu–Al2O3 catalysts prepared by Tsuruya et al. in different methods in the similar catalytic system were 1.25% and 2.5%, respectively.29 Furthermore, in 2006 Gao et al. realized the direct hydroxylation of benzene to phenol by prereducing V–Al2O3 by ascorbic acid before reaction, even though only giving the phenol yield of 4.3%.30 It was found that the V4+ valence vanadium (VO2+) was effective for the reaction. In addition, the V-containing polyoxometalates (POMs) of structure and property diversity were a group of widely studied catalysts in the hydroxylation process.9,31–34 To date, the highest phenol yield of more than 20% could be achieved by the synergy between copper and vanadium oxide species on the support SBA-15.35 Most recently, Han et al. prepared VxOy@C catalysts derived from sucrose and NH4VO3 by hydrothermal method.36 The catalysts showed good catalytic performance for the title reaction using oxygen as oxidant with a phenol yield of 12.2%. Unfortunately, supported vanadium catalysts for oxidation usually suffered from vast leaching of vanadium, which resulting in that the catalysts could not be recycled. Therefore, design and preparation of highly efficient recyclable catalysts for the direct hydroxylation of benzene to phenol still needed further investigation.
As it was reported that N-dopants in the carbon material could strengthen the interaction between the reactive species and supports,37,38 the introduction of N may contribute to overcome the leaching of reactive species. Bearing extreme chemical and thermal stability and easy accessibility of polyaniline (PANI)39 in mind, we selected it as nitrogen and carbon precursor and VO(acac)2 as metal precursor to realize facile one-pot synthesis of vanadium-supported N-doped carbon materials (designated as V–N–C). The use of such a polymer PANI as a nitrogen and carbon precursor promised a more uniform distribution of nitrogen sites on the surface, further resulted in a more uniform distribution of reactive vanadium species. The catalysts were characterized systematically using thermogravimetric analysis (TGA), Fourier transform infrared spectroscopy (FTIR), N2 adsorption–desorption, powder X-ray diffraction (XRD), Raman spectra, X-ray photoelectron spectroscopy techniques (XPS) and scanning electron microscope (SEM). Based on the above characterization, it was found that the special structure of V–N–C catalysts prepared at different pyrolysis temperature varied greatly as well as the chemical state, resulting in different catalytic performance on the direct hydroxylation of benzene to phenol with molecular oxygen as oxidant. Up to 12.6% yield of phenol was achieved with the selectivity as high as 97.8% employing V–N–C-600 catalyst. The influence of reaction conditions, such as reaction time, reaction temperature, and oxygen pressure was investigated. Noteworthily, V–N–C-600 catalyst could be recycled at least six times without significant decline of phenol yield. It was considered that the interaction between surface nitrogen of supports and reactive vanadium species played an important role in the good stability in recycling of the heterogeneous V–N–C catalysts.
2. Results and discussion
2.1. Structure characterization of V–N–C catalysts
The dynamic thermogravimetry analysis (TGA) and the corresponding differential thermogravimetry (DTG) curves during the heat treatment of PANI–V composites were illustrated in Fig. 1. The DTG indicated a multi-stage pyrolysis. At least three features could be distinguished on the DTG curve. One feature appeared as a peak at around 100 °C related to water desorption, if the material was not sufficiently dry. The most important feature was pointed out as a peak in a range of 200 to 400 °C. The highest mass change took place on the DTG trace and could be attributed to the decomposition of the remained acetate ligand. It also meant that the formation of VOx occurred. While the TG and DTG traces up to 500 °C showed no significant change of the PANI–V composites, it could be addressed to the reconstruction and carbonization of carbon nitride matrix PANI to form the N-doped graphene layers. | ||
| Fig. 1 The dynamic TG and the corresponding DTG curves during the heat treatment of PANI–V composites. | ||
Fourier transform infrared (FTIR) spectra of unpyrolyzed PANI–V composites and V–N–C catalysts (Fig. 2) showed that the benzene-type (1170 cm−1) and quinone-type (1400 cm−1) structures40 on the main PANI chain transformed to other structures containing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (1630 cm−1). The weak absorption bands at ca. 1020, 825, and 602 cm−1 of V–N–C catalysts were attributed to the stretching vibrations of isolated VO groups41 and the deformation modes of V–O–V chains.42 Moreover, Fig. S1† illustrated the shape of N2 adsorption–desorption isotherms of V–N–C-600, showing the presence of a certain degree of microporous structure. The textual parameters, including surface areas and pore volumes of V–N–C catalysts were summarized in Table S1.† Increasing the pyrolysis temperature resulted in significant increase in the surface area and pore volume. Meanwhile, the pore size distributions of V–N–C catalysts were relatively broad.
N (1630 cm−1). The weak absorption bands at ca. 1020, 825, and 602 cm−1 of V–N–C catalysts were attributed to the stretching vibrations of isolated VO groups41 and the deformation modes of V–O–V chains.42 Moreover, Fig. S1† illustrated the shape of N2 adsorption–desorption isotherms of V–N–C-600, showing the presence of a certain degree of microporous structure. The textual parameters, including surface areas and pore volumes of V–N–C catalysts were summarized in Table S1.† Increasing the pyrolysis temperature resulted in significant increase in the surface area and pore volume. Meanwhile, the pore size distributions of V–N–C catalysts were relatively broad.
 | ||
| Fig. 2 FT-IR spectra of PANI–V composites and V–N–C catalysts. | ||
The X-ray diffraction (XRD) patterns shown in Fig. 3 displayed the structures of V–N–C catalysts prepared at different temperatures. The broadened peak centers at 25° was observed, corresponding to the (002) plane of carbon material with low graphitization degree.43 By contrast, this characteristic peak exhibited in XRD patterns of V–N–C-600 catalyst was comparatively narrower than that in others, indicating pyrolysis treatment at 600 °C may lead to the formation of more ordered graphite carbon. In addition, the characteristic diffraction peaks of crystalline V2O3 were clearly found in the XRD patterns of V–N–C-1000 catalyst, though with very low crystallinity, which may be attributed to the high-temperature pyrolysis resulting in the formation of crystal phase V2O3. Moreover, Fig. S2† compared Raman spectra of V–N–C catalysts synthesized at different temperatures. As the highly ordered graphite shows a very weak D-band,44 the low ratio of the D-band to G-band integrated intensities proved the formation of more and more ordered graphite structure during the heat treatment at 400, 600 °C, which was consistent with the result of XRD tests.
 | ||
| Fig. 3 XRD patterns of crystallite V2O3 and V–N–C catalysts. | ||
X-ray photoelectron spectroscopy (XPS) measurements were conducted to elucidate the surface chemical composition of V–N–C catalysts. Fig. 4 demonstrated that the chemical composition of V–N–C catalysts was significantly different. As shown in Fig. 4e, it was clear that the increase in the pyrolysis temperature led to a sudden drop in nitrogen content, from 13.24 at% to 2.95 at%. This may be attributed to the breaking of the nitrogen–carbon bond.
 | ||
| Fig. 4 XPS spectra of N 1s (a) V–N–C-400, (b) V–N–C-600, (c) V–N–C-800, (d) V–N–C-1000, (e) gross nitrogen-content of different catalysts, (f) nitrogen configurations for different catalysts. | ||
Nitrogen could be present in different forms of nitrogen doped carbon including pyridinic-N, pyrrolic-N, graphitic-N, quaternary-N, and pyridine-N-oxide. Fig. 4a–d showed the deconvoluted XPS spectra of N 1s of V–N–C catalysts. Meanwhile, three different signals having binding energies of 398.7 eV ± 0.2, 399.8 eV ± 0.2, and 401.0 eV ± 0.2, corresponded to pyridinic N, pyrrolic N, and graphitic N, respectively.45,46 The peak at the binding energy of 398.7 eV ± 0.2, N1, should also include a contribution from nitrogen bound to the metal (N–V), due to the small difference between binding energies of N–metal and pyridinic N.46,47 Pyridinic N (probably including N–V) and graphitic N are generally believed to participate in the activation of molecular oxygen.46–48 The N1 content increased to the maximum as the pyrolysis temperature increased from 400 to 600 °C, and then immediately began to gradually decrease with the continue increasing in the pyrolysis temperature (Fig. 4f). The pyrrolic-N (399.8 eV ± 0.2, N2) refers to the N atoms bonded with two carbon atoms, which is incorporated into five member heterocyclic rings. The N2 content quickly decreased as the pyrolysis temperature increased, which indicated that the N1 was more stable than the N2. As we know, quaternary-N (401.0 eV, N3) is the graphitic nitrogen in the centre of the heterocyclic rings, which is located in the perfect graphite plane by bonding with three carbon atoms. However, for the peak at 401.9 eV ± 0.2, N4, some researchers believed that it was contributed to the pyridine-N-oxide49 and some thought it reflected the presence of graphitic-N at the valley, which is located at the edge of the heterocyclic rings.39,50,51 It had been reported that the nitrogen oxide functionalities exhibited low thermal stability.49 Indeed, the ratio of N4 increased with the increase in the pyrolysis temperature. Therefore, this peak in this article should be assigned to graphite nitrogen, which was in agreement with the observations of Sharifi et al.51 Interestingly, the perfect graphite N3 ratio sharply decreased with the increase to 800 °C in the pyrolysis temperature, which mainly attributed to the sudden increase of the N4 ratio. It indicated that the structure of perfect graphite nitrogen-doped carbon structure transformed into other forms. Moreover, pyridine-N-oxide (403.5 ± 0.2 eV, N5) refers to N atom bonded with two carbon atoms and one oxygen atom. The N5 content fluctuated in some degree as the temperature increased.
From the XPS data, the O 1s profile could be separated into three peaks (Fig. 5). That was, there existed three types of O species, carbonyl groups, alcohol and/or ether groups, and carboxyl and/or ester groups at 531.6, 532.5 and 533.5 eV, respectively.52 The XPS spectra of V–N–C catalysts showed the binding energy of V 2p1/2 at 523.4 eV and the binding energy of V 2p3/2 in the range of 515.5–517.0 eV,30,53 indicating the existence of V4+ and V5+ on the surface of different catalysts, but no V3+ (515.1 eV). It was because the surface V3+ is easily oxidized to V4+ when exposed to air. Additionally, low content and amorphous phase of V4+ and V5+ resulted in that no corresponding diffraction peaks existed in the XRD patterns of V–N–C catalysts. Furthermore, Fig. 5e showed that the surface V contents of V–N–C catalysts were 0.83, 0.67, 0.62 and 1.04 wt% for V–N–C-400, V–N–C-600, V–N–C-800 and V–N–C-1000 catalysts, respectively. As shown in Fig. 5f, the ratio of surface V4+ of V–N–C-600 catalyst was comparatively higher, while that of V–N–C-1000 with the highest surface gross vanadium was lowest among them. The surface morphology of V–N–C-600 catalyst was determined by SEM (Fig. S3†). Furthermore, the VOx particles were uniformly incorporated in the nitrogen-doped carbon supports as shown in Fig. S4a and b.† Moreover, as illustrated by energy-dispersive X-ray spectroscopy (EDXS) analysis (Fig. S4c†), the particles contained vanadium.
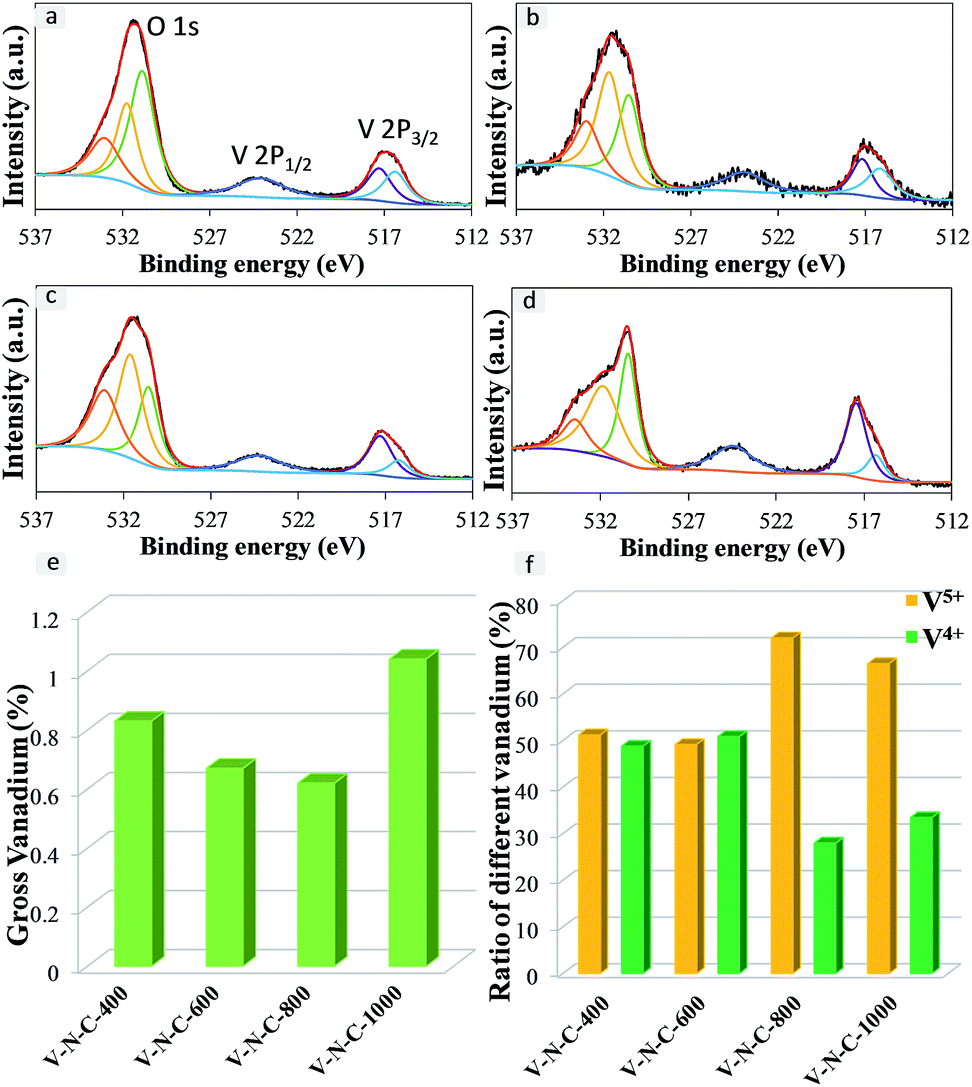 | ||
| Fig. 5 XPS spectra of O 1s, V 2p (a) V–N–C-400, (b) V–N–C-600, (c) V–N–C-800, (d) V–N–C-1000, (e) gross vanadium-content of different catalysts, (f) vanadium configurations for different catalysts. | ||
2.2. Catalytic performances of V–N–C catalysts for the direct hydroxylation of benzene to phenol
A series of V–N–C catalysts were prepared by pyrolyzing PANI–V composites under different temperature. ICP tests indicated that V content in the catalysts increased gradually with the increase of the pyrolysis temperature, which may due to high temperature treatment resulted in a certain amount of loss of carbon nitride supports. Furthermore, the catalytic performances of V–N–C catalysts for the direct hydroxylation of benzene to phenol with molecular oxygen were evaluated. In all reactions, phenol and hydroquinone were produced without observing the formation of catechol and benzoquinone. As shown in Table 1, trace phenol was obtained in the absence of catalyst (Table 1, entry 1). Ascorbic acid was used as the sacrificial reductant, which proved to be essential for the hydroxylation reaction (Table 1, entry 2). To our delight, the yield of phenol first increased with pyrolysis temperature and then decreased with a threshold of 600 °C, giving 11.8% yield with 97.0% selectivity (Table 1, entries 3–6). Though not significant rangeability which may result from the inert reactivity of benzene, it was understandable considering that V–N–C-600 catalyst with a higher ratio of surface V4+ active species performed better than others. On the contrary, V–N–C-1000 catalyst with a lower ratio of surface V4+ only obtained 9.2% yield with 95.6% selectivity, even though the surface gross vanadium was higher than others (Table 1, entry 6), which was consistent with that V4+ have been proved to be more active for activating oxygen.36,54 In addition, it could be speculated that specific surface area and specific volume of catalysts are not the only factor affecting the catalytic performance in consideration of the highest specific surface area and specific volume of V–N–C-1000 catalyst. Moreover, only trace phenol was obtained when N–C-600 catalyst was used as catalyst, which proved that V species were necessary for the direct hydroxylation of benzene to phenol with molecular oxygen as oxidant (Table 1, entry 7). Next we studied the influence of reaction time on the direct hydroxylation of benzene to phenol over V–N–C-600 catalyst. When the reaction time reduced to 6 h, only 7.0% yield of phenol was obtained with 99.5% selectivity (Table 1, entry 8). When the reaction time was 12 h, 9.6% yield of phenol was produced with up to 99.9% selectivity (Table 1, entry 9). When the reaction time was further prolonged to 36 h, the yield of phenol was increased to 12.6% with the selectivity slightly decreased to 97.8% (Table 1, entry 10), which was higher than that reported in the literature with acetonitrile as solvent and molecular oxygen as oxidant.33,36| Entry | Catalysta | V% (ICP) | Yieldb (%) | Sel.b (%) |
|---|---|---|---|---|
| a Reaction conditions: 10 mmol benzene, 1.4 mol% V, 0.88 g ascorbic acid, 2 g acetonitrile, 2 MPa O2, 80 °C, 24 h.b Determined by GC.c Without an addition of catalysts.d No by-product was detected.e Without ascorbic acid.f 6 h.g 12 h.h 36 h. | ||||
| 1c | None | 0 | Trace | —d |
| 2e | V–N–C-600 | 4 | Trace | — |
| 3 | V–N–C-400 | 4.3 | 10.1 | 98.7 |
| 4 | V–N–C-600 | 4.0 | 11.8 | 97.0 |
| 5 | V–N–C-800 | 6.4 | 10.8 | 97.0 |
| 6 | V–N–C-1000 | 9.5 | 9.2 | 95.6 |
| 7 | N–C-600 | 0 | Trace | — |
| 8f | V–N–C-600 | 4.0 | 7.0 | 99.5 |
| 9g | V–N–C-600 | 4.0 | 9.6 | 99.9 |
| 10h | V–N–C-600 | 4.0 | 12.6 | 97.8 |
Fig. 6 depicted the influence of reaction temperature on the direct hydroxylation of benzene to phenol over V–N–C-600 catalyst. The yield of phenol first increased as the reaction temperature increased to 80 °C and then decreased. The decrease of the oxygen solubility with an increase of temperature was one of the main reasons for this phenomenon.55 The self-oxidation of ascorbic acid as the sacrificial reductant prolonged the reaction,29 which also caused the decline in the yield of phenol. Additionally, it was found when the reaction temperature was higher than 80 °C, the selectivity of phenol sharply decreased, indicating that overoxidation of benzene prefers higher reaction temperature.
 | ||
| Fig. 6 The influence of reaction temperature on the direct hydroxylation of benzene to phenol over V–N–C-600 catalyst. The yield (column) and selectivity (■) of phenol. Reaction conditions: 10 mmol benzene, 1.4 mol% V, 0.88 g ascorbic acid, 2 g acetonitrile, 2 MPa O2, 12 h. | ||
The influence of oxygen pressure was also investigated (Fig. 7). In the low pressure range, the yield of phenol was enhanced to a maximum 10.7% with the oxygen pressure increased to 3.0 MPa, which may be attributed to the larger solubility of oxygen at the higher oxygen pressure.55 However, the selectivity of phenol significantly decreased at the same time. Especially, when the oxygen pressure further increased, the selectivity and yield of phenol were both obviously decreased. It was believed that too much oxygen might cause the overoxidation of phenol, which resulted in a decrease in the selectivity and yield of phenol.
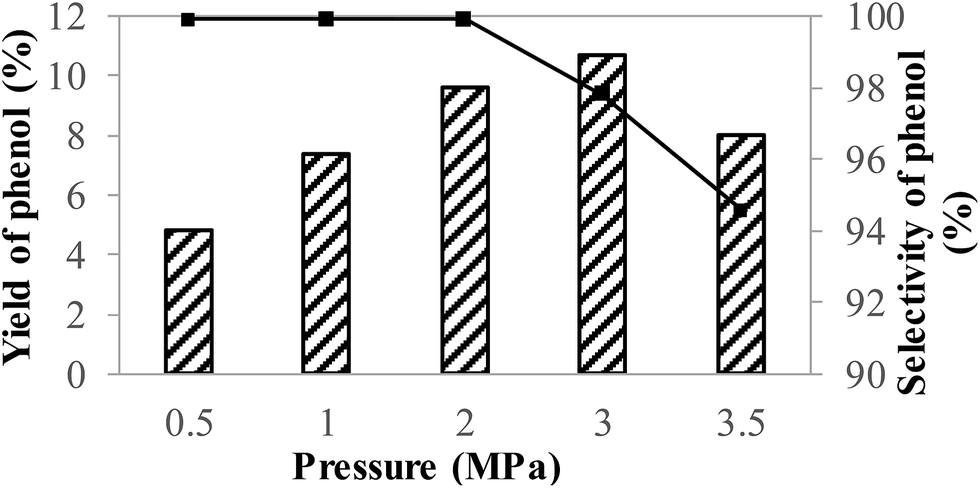 | ||
| Fig. 7 The influence of oxygen pressure on the direct hydroxylation of benzene to phenol over V–N–C-600 catalyst. The yield (column) and selectivity (■) of phenol. Reaction conditions: 10 mmol benzene, 1.4 mol% V, 0.88 g ascorbic acid, 2 g acetonitrile, 80 °C, 12 h. | ||
The efficacy of a heterogeneous catalyst should also be evaluated from its recyclability and stability. V–N–C-600 catalyst presented considerable recyclability towards the phenol formation with the above 98% selectivity over six catalytic cycles, as shown in Fig. 8. The yield of phenol decreased during the first running cycle, while it obviously improved by calcining the recycled catalyst under flowing nitrogen at 400 °C for 2 hours. It could be speculated that one possible reason accounting for the phenomenon was the absorption of substrate, product and high-boiling byproduct for the catalyst. Afterwards, the recycled catalyst presented nice recyclability, but a slight decrease compared with the first test, which may be due to the leaching of unstable V species (leaching of V after the first run: 17.9%, determined by ICP). Moreover, compared with fresh V–N–C-600 catalyst, the FTIR spectra, XRD patterns and V XPS spectra of the reactivated catalyst after sixth run did not show any obvious change, as shown in Fig. S5 and S6.† Considerable recyclability of V–N–C-600 catalyst could be ascribed to the presence of the interaction of nitrogen and vanadium, which avoided the vast leaching of V, generally existing in the supported vanadium catalytic oxidation.
 | ||
| Fig. 8 Reuse of the catalysts for the direct hydroxylation of benzene to phenol. The yield (column) and selectivity (■) of phenol. Reaction conditions: 10 mmol benzene, 1.4 mol% V, 0.88 g ascorbic acid, 2 g acetonitrile, 2 MPa O2, 80 °C, 12 h. | ||
3. Experimental
3.1. Materials
Aniline, acetonitrile, 1,4-dioxane, ammonium persulfate (98.0%) and ascorbic acid were commercially available. Vanadyl(IV) acetylacetonate (99%) was obtained from Acros Organics (China). The above reagents were used as purchased without further purification unless other noted.3.2. Catalyst preparation
Briefly, 1000 mg aniline monomer and 265.2 mg VO(acac)2 were dissolved in 20 mL 1.0 M HCl successively. After stirring for 10 minutes, 5 mL 1.0 M HCl solution containing 2.5 g ammonium peroxydisulfate (APS) was added dropwise with vigorous stirring. The polymerization was conducted in an ice bath (<5 °C) for 24 h. After evaporation of water at 100 °C, the obtained dark-green PANI–V composites were then pyrolyzed under flowing nitrogen at different temperature for 2 h. After calcination, the dark sample was further cooled under nitrogen until to the room temperature. The obtained catalysts were designated as V–N–C.For comparison, N–C-600 catalyst was also prepared as the above process but without addition of VO(acac)2.
After the reaction, the reaction mixture was centrifuged and the solid catalysts were recovered, following by washing with ethanol at least three times. The obtained recycled catalysts were calcined under flowing nitrogen at 400 °C for 2 hours, and then reused in the next run.
3.3. Catalyst activity test
The hydroxylation of benzene was performed in a Teflon-lined stainless-steel reactor (40 mL) that was equipped with a pressure gauge, thermocouple, gas-inlet valve, magnetic stirrer, and an electric heater with a controller. In a typical experiment, 175 mg of the catalyst, 0.88 g of ascorbic acid, and 10 mmol of benzene were added to 2.0 g of acetonitrile successively. After the system was charged with 2.0 MPa of O2 at room temperature, the hydroxylation reaction was conducted at 80 °C for the desired time under vigorous stirring. After the reaction, 1,4-dioxane was added into the product mixture as an internal standard for product analysis. The mixture was analyzed using a gas chromatograph (GC) of Agilent 7890 equipped with a flame ionization detector (FID). The products of phenol and hydroquinone were detected using GC in this reaction system. The yield of phenol was calculated as: (mole of formed phenol)/(mole of initial benzene) × 100%.3.4. Catalyst characterization
Thermogravimetric analysis (TGA) and the corresponding differential thermogravimetry (DTG) of the samples were conducted by using a Perkin-Elmer TGA-2 thermo gravimetric analyzer in N2 from room temperature to 800 °C at a rate of 10 °C min−1. The V loadings of the catalysts were measured by inductively coupled plasma atomic emission spectroscopy (ICP-AES) using a Perkin-Elmer OPTIMA 3300DV. The detection limit was 0.10 ppm. N2 adsorption–desorption isotherms were determined at −196 °C using a QuadraSorb SI4 Station. Before measurement, the samples were degassed in vacuum at 300 °C for 3 h, and the BET surface areas of the samples were calculated using adsorption data. The crystal structure of V–N–C were characterized with power X-ray diffraction (XRD) patterns on a Rigaku D/Max 2500PC diffractometer equipped with a Cu Ka radiation source (λ = 1.5418 Å) at a scanning rate of 0.05° s−1 (2θ from 20° to 75°). Visible Raman spectra were recorded at room temperature on a Jobin Yvon LabRAM HR 800 instrument with a 532 nm excitation laser at a power of around 20 mW. Surface composition was determined by X-ray photoelectron spectroscopy (XPS) using Thermo Scientific ESCALAB 250Xi instrument with Al-Ka radiation anode (hν = 1486.6 eV). The sample was pressed into a sample holder, evacuated in a load lock to 10−8 Pa, and transferred to the analysis chamber. The C 1s line (284.6 eV) was used as the reference to correct the banding energies (BE). Scanning electron microscope (SEM) image was conducted On a JSM-7800F microscope operated at 20 kV landing energy.4. Conclusion
A series of vanadium-supported N-doped carbon materials (V–N–C) had been synthesized by using polyaniline as the precursor through a facile one-pot pyrolysis method. These catalysts were active for catalyzing the hydroxylation of benzene with molecular oxygen as the oxidant under mild conditions. Especially, V–N–C-600 catalyst with the higher ratio of V4+ was more effective among these catalysts. The reaction time, reaction temperature and oxygen pressure had significant effects on the yield of phenol. A phenol yield of up to 12.6% could be obtained with a selectivity of 97.8%. The V–N–C-600 catalyst was also stable and could be recycled at least six times without significant decline in yield.Acknowledgements
The authors thank the financial support from the National Natural Science Foundation of China (Grant no. 21273225).Notes and references
- J. H. Tyman, Synthetic and natural phenols, Elsevier, 1996 Search PubMed.
- M. Weber, M. Weber and M. Kleine-Boymann, Ullmann's Encyclopedia of Industrial Chemistry, 2004 Search PubMed.
- T. Jiang, W. Wang and B. Han, New J. Chem., 2013, 37, 1654–1664 RSC.
- K. Weissermehl and H. Arpe, Industrial Organische Chemie, VCH, Weinheim, 1988 Search PubMed.
- B. Cornils and W. A. Herrmann, J. Catal., 2003, 216, 23–31 CrossRef CAS.
- J. L. Smith and R. Norman, J. Chem. Soc., 1963, 2897–2905 RSC.
- N. Herron and C. A. Tolman, J. Am. Chem. Soc., 1987, 109, 2837–2839 CrossRef CAS.
- Y. Liu, K. Murata and M. Inaba, J. Mol. Catal. A: Chem., 2006, 256, 247–255 CrossRef CAS PubMed.
- Z. Long, Y. Zhou, G. Chen, W. Ge and J. Wang, Sci. Rep., 2014, 4, 3651 CAS.
- S. Shang, H. Yang, J. Li, B. Chen, Y. Lv and S. Gao, ChemPlusChem, 2014, 79, 680–683 CrossRef CAS.
- C. Walling and R. A. Johnson, J. Am. Chem. Soc., 1975, 97, 363–367 CrossRef CAS.
- H. Mimoun, L. Saussine, E. Daire, M. Postel, J. Fischer and R. Weiss, J. Am. Chem. Soc., 1983, 105, 3101–3110 CrossRef CAS.
- M. L. Neidig and K. F. Hirsekorn, Catal. Commun., 2011, 12, 480–484 CrossRef CAS PubMed.
- P. Borah, X. Ma, K. T. Nguyen and Y. Zhao, Angew. Chem., Int. Ed., 2012, 51, 7756–7761 CrossRef CAS PubMed.
- P. Zhao, J. Wang, G. Chen, Y. Zhou and J. Huang, Catal. Sci. Technol., 2013, 3, 1394–1404 CAS.
- G. Ding, W. Wang, T. Jiang, B. Han, H. Fan and G. Yang, ChemCatChem, 2013, 5, 192–200 CrossRef CAS.
- J.-H. Yang, G. Sun, Y. Gao, H. Zhao, P. Tang, J. Tan, A.-H. Lu and D. Ma, Energy Environ. Sci., 2013, 6, 793–798 CAS.
- J. Xu, Q. Jiang, T. Chen, F. Wu and Y.-X. Li, Catal. Sci. Technol., 2015, 447–454 CAS.
- M. Iwamoto, J. Hirata, K. Matsukami and S. Kagawa, J. Phys. Chem., 1983, 87, 903–905 CrossRef CAS.
- V. Sobolev, A. Kharitonov, Y. A. Paukshtis and G. Panov, J. Mol. Catal., 1993, 84, 117–124 CrossRef CAS.
- R. Leanza, I. Rossetti, I. Mazzola and L. Forni, Appl. Catal., A, 2001, 205, 93–99 CrossRef CAS.
- J. Jia, K. S. Pillai and W. M. Sachtler, J. Catal., 2004, 221, 119–126 CrossRef CAS PubMed.
- V. S. Chernyavsky, L. V. Pirutko, A. K. Uriarte, A. S. Kharitonov and G. I. Panov, J. Catal., 2007, 245, 466–469 CrossRef CAS PubMed.
- S. Niwa, M. Eswaramoorthy, J. Nair, A. Raj, N. Itoh, H. Shoji, T. Namba and F. Mizukami, Science, 2002, 295, 105–107 CrossRef CAS PubMed.
- K. LunáYeung, Chem. Commun., 2009, 5898–5900 Search PubMed.
- X. Wang, X. Tan, B. Meng, X. Zhang, Q. Liang, H. Pan and S. Liu, Catal. Sci. Technol., 2013, 3, 2380–2391 CAS.
- T. Ohtani, S. Nishiyama, S. Tsuruya and M. Masai, J. Catal., 1995, 155, 158–162 CrossRef CAS.
- J. Okamura, S. Nishiyama, S. Tsuruya and M. Masai, J. Mol. Catal. A: Chem., 1998, 135, 133–142 CrossRef CAS.
- T. Miyahara, H. Kanzaki, R. Hamada, S. Kuroiwa, S. Nishiyama and S. Tsuruya, J. Mol. Catal. A: Chem., 2001, 176, 141–150 CrossRef CAS.
- X. Gao and J. Xu, Catal. Lett., 2006, 111, 203–205 CrossRef CAS PubMed.
- S. Sumimoto, C. Tanaka, S. T. Yamaguchi, Y. Ichihashi, S. Nishiyama and S. Tsuruya, Ind. Eng. Chem. Res., 2006, 45, 7444–7450 CrossRef CAS.
- H. Ge, Y. Leng, C. Zhou and J. Wang, Catal. Lett., 2008, 124, 324–329 CrossRef CAS.
- H. Yang, J.-Q. Chen, J. Li, Y. Lv and S. Gao, Appl. Catal., A, 2012, 415, 22–28 CrossRef PubMed.
- H. Yang, J. Li, L. Wang, W. Dai, Y. Lv and S. Gao, Catal. Commun., 2013, 35, 101–104 CrossRef CAS PubMed.
- Y.-Y. Gu, X.-H. Zhao, G.-R. Zhang, H.-M. Ding and Y.-K. Shan, Appl. Catal., A, 2007, 328, 150–155 CrossRef CAS PubMed.
- W. Wang, G. Ding, T. Jiang, P. Zhang, T. Wu and B. Han, Green Chem., 2013, 15, 1150–1154 RSC.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- R. Nie, J. Shi, W. Du, W. Ning, Z. Hou and F.-S. Xiao, J. Mater. Chem. A, 2013, 1, 9037–9045 CAS.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- M. Baibarac, I. Baltog, S. Lefrant, J. Mevellec and O. Chauvet, Chem. Mater., 2003, 15, 4149–4156 CrossRef CAS.
- S. B. Kristensen, A. J. Kunov-Kruse, A. Riisager, S. B. Rasmussen and R. Fehrmann, J. Catal., 2011, 284, 60–67 CrossRef CAS PubMed.
- N. Pinna, M. Willinger, K. Weiss, J. Urban and R. Schlögl, Nano Lett., 2003, 3, 1131–1134 CrossRef CAS.
- L. Lin, Q. Zhu and A.-W. Xu, J. Am. Chem. Soc., 2014, 136, 11027–11033 CrossRef CAS PubMed.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef CAS PubMed.
- Y. Nabae, S. Moriya, K. Matsubayashi, S. M. Lyth, M. Malon, L. Wu, N. M. Islam, Y. Koshigoe, S. Kuroki and M.-a. Kakimoto, Carbon, 2010, 48, 2613–2624 CrossRef CAS PubMed.
- U. Koslowski, I. Herrmann, P. Bogdanoff, C. Barkschat, S. Fiechter, N. Iwata, H. Takahashi and H. Nishikori, ECS Trans., 2008, 13, 125–141 CAS.
- G. Wu, C. M. Johnston, N. H. Mack, K. Artyushkova, M. Ferrandon, M. Nelson, J. S. Lezama-Pacheco, S. D. Conradson, K. L. More and D. J. Myers, J. Mater. Chem., 2011, 21, 11392–11405 RSC.
- Y. Zhao, K. Watanabe and K. Hashimoto, J. Am. Chem. Soc., 2012, 134, 19528–19531 CrossRef CAS PubMed.
- S. Kundu, T. C. Nagaiah, W. Xia, Y. Wang, S. V. Dommele, J. H. Bitter, M. Santa, G. Grundmeier, M. Bron and W. Schuhmann, J. Phys. Chem. C, 2009, 113, 14302–14310 CAS.
- H. Peng, Z. Mo, S. Liao, H. Liang, L. Yang, F. Luo, H. Song, Y. Zhong and B. Zhang, Sci. Rep., 2013, 3, 1765 Search PubMed.
- T. Sharifi, G. Hu, X. Jia and T. Wågberg, ACS Nano, 2012, 6, 8904–8912 CrossRef CAS PubMed.
- J. Xu, H. Liu, R. Yang, G. Li and C. Hu, Chin. J. Catal., 2012, 33, 1622–1630 CrossRef CAS.
- Y. He, Y. Wang, L. Zhao and X. Wu, J. Mol. Catal. A: Chem., 2011, 337, 61–67 CrossRef CAS PubMed.
- Y.-k. Masumoto, R. Hamada, K. Yokota, S. Nishiyama and S. Tsuruya, J. Mol. Catal. A: Chem., 2002, 184, 215–222 CrossRef CAS.
- S. T. Yamaguchi, S. Sumimoto, Y. Ichihashi, S. Nishiyama and S. Tsuruya, Ind. Eng. Chem. Res., 2005, 44, 1–7 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra04836b |
| This journal is © The Royal Society of Chemistry 2015 |