
One-pot synthesis of a TiO2–CdS nano-heterostructure assembly with enhanced photocatalytic activity†
Abstract
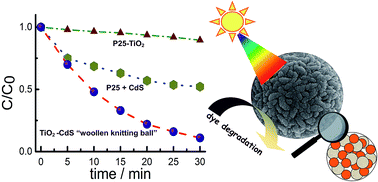
An unprecedented morphology of a titanium dioxide (TiO2) and cadmium sulfide (CdS) self-assembly obtained using a ‘truly’ one-pot and highly cost effective method with a multi-gram scale yield is reported here. The TiO2–CdS assembly, comprising of TiO2 and CdS nanoparticles residing next to each other homogeneously self-assembling into ‘woollen knitting ball’ like microspheres, exhibited remarkable potential as a visible light photocatalyst with high recyclability.
 Please wait while we load your content...
Please wait while we load your content...

