DOI:
10.1039/C5RA04685H
(Paper)
RSC Adv., 2015,
5, 36022-36029
A first principles study of the mechanical properties of Li–Sn alloys
Received
17th March 2015
, Accepted 14th April 2015
First published on 14th April 2015
Abstract
To obtain a better understanding of the failure mechanism of lithium ion batteries during charging–discharging, we have systematically studied the mechanical properties of Li–Sn alloys based on density functional theory, including elastic constants such as the orientation-averaged bulk, shear and Young's moduli and Poisson's ratios, as well as the anisotropy and brittleness-ductility. It is shown that bulk, shear and Young's moduli of isotropic LixSn alloys decrease almost linearly with the increase of Li concentration. Further, based on the analysis of shear to bulk modulus and Poisson's ratios, it is inferred that Li–Sn alloys are brittle. The poor cycle performance and crushing failure of Sn anode materials during charging and discharging are mainly due to a transition of electrode material properties from ductile to brittle.
1. Introduction
Over the last decade, the demand for high-energy storage has been rapidly increasing. There are more and more studies focusing on new active materials of batteries with high voltage, large capacity and high rate capability.1,2 As an alternative energy storage device, lithium ion batteries (LIBs) have become the most promising portable power sources in consumer electronics and vehicle electrification. Here, one of the most critical parameters for the next generation of LIBs is the energy density or specific energy.3 As is well known, LIBs can store more energy per weight than that of their competitors such as nickel–metal hydride, nickel–cadmium and lead-acid batteries.
Up to now, graphite with a layer structure is the most widely used negative electrode material in LIBs, and during charging, the compound LiC6 is formed from the lithium–graphite interaction. Unfortunately, its theoretical capacity is relatively low (372 mA h g−1).4,5 In contrast, Sn exhibits an extraordinarily large theoretical capacity of 994 mA h g−1, which is about two and a half times of the conventional graphite anode. As an anode material, however, Sn experiences a massive volume change (∼300%) during a charge–discharge process, and binary alloys are formed during lithiation, e.g., Li2Sn5, LiSn, Li7Sn3, Li5Sn2, Li13Sn5, Li7Sn2 and Li17Sn4.6 Thus, the large volume expansion, phase transition and Li diffusion-induced stress within electrode materials may result in their capacity loss and poor cycling performance due to electrode damage and fracture.7
To improve the durability of LIBs, significant progresses have been made in understanding the Li ion diffusion-induced stress. For example, under galvanostatic or potentiostatic operation, the stress and strain evolution within a spherical particle was modeled.8 The effects of surface energy and elasticity on the stress evolution in spherical electrodes were also investigated.9 Wolfenstine demonstrated that a critical particle size for lowering the capacity fade can be estimated by using Young's modulus, fracture toughness and volume deformation.10 It is also shown that, upon lithiation, the change of the averaged Young's modulus of LixFePO4 is less than 1%.11 However, Young's moduli of crystalline and amorphous LixSi electrodes decrease almost linearly with Li concentration, and when forming the phase Li15Si4, Young's modulus drops to one-third of its original value.12 Thus, it is necessary to have a better understanding on the Li concentration-dependent mechanical properties of LIBs.
In this paper, we calculated the elastic constants of Li–Sn alloys based on the density functional theory. These constants such as bulk, shear and Young's moduli and Poisson's ratios can be obtained with the help of the Voight–Reuss–Hill approximation.13 Other properties of Li–Sn alloys, including the anisotropy and brittleness-ductility, were also calculated. Finally, it is shown that there is a quantitative relationship between the mechanical properties of Li–Sn alloys and Li concentration, which is important for evaluating deformation and failure of electrode materials.
2. Calculation methodology
2.1 Structure optimization
By using the Vienna Ab initio Simulation Package with the local density approximation for exchange and correction, we calculated the structural and elastic properties of Li–Sn alloys based on the density functional theory. The projected augmented wave method was used to describe the ion–electron interaction.14,15 Here, valence configurations for Li and Sn are 1s22s1 and 5s25p2 (4d10), respectively. The kinetic energy cutoff for the plane wave basis set was converged for every structure and its value in the plane wave expansion is 500 eV. In calculations, all atoms were fully relaxed by using the conjugate gradient method until residual forces on constituent atoms were less than 5 × 10−2 eV Å−1, and in determining mechanical properties, the value of convergent force increases to 1 × 10−3 eV Å−1. In addition, the Brillouin zone was sampled with the Monkhorst-Pack k-point grid.16
2.2 Elastic constants
By applying a small strain to the equilibrium lattice, the elastic constants, Cij, in a crystalline system can be deduced from the change of its total energy in a unit cell. The strain tensor εij has six independent components, and with the Voigt notation, they can be expressed as| |
 | (1) |
Under a small strain εij, the relationship between the strain energy and stress σij is given by
| |
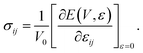 | (2) |
According to the generalized Hooke law, σij = Cijklεkl, the elastic constants can be identified as the second derivative of the total energy to strain, that is
| |
 | (3) |
Here, the total energy of a crystal is expanded in the following Taylor form:
| |
 | (4) |
where
V0 is the volume of the unstrained system and
E(
V0, 0) is the corresponding total energy.
Then, through selecting a specific strain εij = (e1, e2, e3, e4, e5, e6), we can determine the total energy change before and after a set of different strains (≤2%) and quadratic coefficients. According to the Voight–Reuss–Hill approximation,13 bulk and shear moduli, K and G, can be calculated by
and
where subscripts ‘V’ and ‘R’ indicate the Voight and Reuss approximations, respectively.
KV,
KR,
GV and
GR are defined as
| |
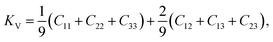 | (7) |
| |
 | (8) |
| |
 | (9) |
and
| |
 | (10) |
where
Sij are the elastic compliance constants,
i.e., the inverse matrix of elastic constants. Then, Young's modulus
Y and Poisson's ratio
ν can be obtained by
and
| | |
ν = (3K − 2G)/2(3K + G)
| (12) |
3. Results and discussion
3.1 Structure properties
Structure optimization is firstly performed to determine the lattice parameters and the positions of atoms in Li–Sn alloys. To obtain an equilibrium volume, all structures are fully relaxed. As is well known, the local density approximation usually underestimates lattice constants and overestimates elastic constants, and while the generalized gradient approximation is just opposite. Thus, we use the local density approximation for exchange-correction, and due to an over-binding problem of the local density approximation, it is uniform in estimating all structures. The structure data of crystalline Li–Sn phases obtained from calculations are summarized in Tables 1 and 2, and for comparison, the available experimental data are also listed (see Table 2). It is obvious that our results are well agreement with experimental data.
Table 1 Crystal structure information for Li, Sn and LixSn, where y is the ratio of Li atoms to the total number of atoms in alloys
| Phase |
Structure |
x |
y = x/(1 + x) |
Space group |
| β-Sn |
 |
0.00 |
0.000 |
I41/amd |
| Li2Sn5 |
 |
0.40 |
0.286 |
P4/mbm |
| LiSn |
 |
1.00 |
0.500 |
P2/m |
| Li7Sn3 |
 |
2.33 |
0.700 |
P21/m |
| Li5Sn2 |
 |
2.50 |
0.714 |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m m |
| Li13Sn5 |
 |
2.60 |
0.722 |
Pm1 |
| Li7Sn2 |
 |
3.50 |
0.778 |
Cmmm |
| Li17Sn4 |
 |
4.25 |
0.810 |
F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m 3m |
| Li |
 |
∞ |
1.000 |
Im3m |
Table 2 Lattice constants in units of Å and k-points used in the calculation, where the experimental values are listed in parentheses
| Phase |
a |
b |
c |
Volume |
k-points |
| β-Sn |
5.792(5.831) |
5.792(5.831) |
3.122(3.184)32 |
104.78 |
9 × 9 × 16 |
| Li2Sn5 |
10.331(10.274) |
10.331(10.274) |
3.169(3.125)33 |
338.20 |
3 × 3 × 11 |
| LiSn |
5.162(5.172) |
7.764(7.742) |
3.233(3.182)34 |
125.00 |
7 × 10 × 4 |
| Li7Sn3 |
9.495(9.451) |
8.536(8.561) |
4.738(4.721)35 |
369.00 |
4 × 7 × 4 |
| Li5Sn2 |
4.725(4.740) |
4.725(4.740) |
119.844(19.833)36 |
383.75 |
8 × 8 × 2 |
| Li13Sn5 |
4.703(4.701) |
4.703(4.701) |
17.130(17.124)37 |
328.14 |
8 × 8 × 2 |
| Li7Sn2 |
9.847(9.802) |
13.838(13.803) |
4.712(4.752)38 |
642.00 |
3 × 2 × 7 |
| Li17Sn4 |
13.907(19.690) |
13.907(19.690) |
13.907(19.690)39 |
7608.27 |
2 × 2 × 2 |
| Li |
3.436(3.513) |
3.436(3.513) |
3.436(3.513)40 |
40.58 |
22 × 22 × 22 |
3.2 Elastic properties
Different deformation modes are applied to obtain the elastic constants of various crystal systems. For example, for tetragonal Li2Sn5 and β-Sn phases, six deformation modes are considered to determine C11, C12, C13, C33, C44 and C66.17 Similarly, five deformation modes for hexagonal Li–Sn phases are used to calculate C11, C12, C13, C33 and C44.18 In order to obtain nine independent elastic constants of orthorhombic Li7Sn2, we apply three monoclinic strain tensor, three orthorhombic strain tensor and expansion along three high-symmetry directions.19 For monoclinic LiSn and Li7Sn3 that have thirteen independent elastic constants, we apply the same nine strain tensors performed on the orthorhombic phase and an additional four.20 All the calculated elastic constants are given in Table 3. In the cases of tetragonal structure β-Sn and cubic structure Li, the calculated elastic constants are C11 = 77.93, C33 = 94.94, C44 = 20.38, C66 = 25.82, C12 = 55.70, C13 = 46.52 and C11 = 16.03, C12 = 13.07, C44 = 11.84, respectively. They are consistent with their corresponding experimental data such as C11 = 73.4, C33 = 90.7, C44 = 21.9, C66 = 25.82, C12 = 59.9, and C13 = 39.1 for β-Sn and C11 = 14.39, C12 = 12.04, and C44 = 11.58 for Li.21,22
Table 3 Elastic constants Cij for Li, Sn and LixSn alloys, where all quantities are in units of GPa
| Phase |
C11 |
C22 |
C33 |
C44 |
C55 |
C66 |
C12 |
C13 |
C23 |
| β-Sn |
77.92 |
77.92 |
94.94 |
20.38 |
20.38 |
25.58 |
55.70 |
46.52 |
46.52 |
| Li2Sn5 |
77.29 |
77.29 |
102.47 |
30.55 |
30.55 |
37.89 |
41.77 |
16.62 |
16.62 |
| LiSn |
57.70 |
87.36 |
58.42 |
16.20 |
15.20 |
17.35 |
14.96 |
26.56 |
21.32 |
| Li7Sn3 |
56.26 |
76.17 |
80.53 |
10.26 |
37.20 |
35.48 |
18.00 |
18.03 |
−6.50 |
| Li5Sn2 |
74.81 |
74.81 |
109.89 |
17.66 |
17.66 |
30.36 |
14.08 |
−4.46 |
−4.46 |
| Li13Sn5 |
75.12 |
75.12 |
101.15 |
20.11 |
20.11 |
31.34 |
12.45 |
−3.20 |
−3.20 |
| Li7Sn2 |
53.53 |
63.83 |
74.23 |
10.62 |
33.38 |
35.23 |
19.74 |
11.20 |
−4.13 |
| Li17Sn4 |
48.30 |
48.30 |
48.30 |
23.78 |
23.78 |
23.78 |
15.41 |
15.41 |
15.41 |
| Li |
16.03 |
16.03 |
16.03 |
11.84 |
11.84 |
11.84 |
13.07 |
13.07 |
13.07 |
Based on these elastic constants, other mechanical parameters for Li–Sn alloys, such as K, G, Y, and ν, can be derived by the Voight–Reuss–Hill approximation.13 The orientation-averaged elastic constants and ν for crystalline Li–Sn phases are plotted in Fig. 1. Here, the Li fraction y is defined as the ratio of Li atoms (x) in LixSn alloys, i.e., y = x/(1 + x). Obviously, such a dimensionless variable y for all the alloy phases ranges from 0 (Sn) to 1 (Li), which allows us to assess the validity of the law of mixtures.
 |
| | Fig. 1 (a) Bulk modulus (K), Young's modulus (Y), and shear modulus (G) and (b) Poisson's ratio (ν) versus Li fraction y. | |
As shown in Fig. 1, K, G, and Y as well as ν decrease almost linearly with the increase of Li concentration, demonstrating a weakening tendency of the resistant ability of Li–Sn alloys to deformation. This is because the Sn–Sn covalent bonds are replaced by the weaker Li–Sn and Li–Li bonds during lithiation, leading to a weaker average strength of constituent chemical bonds in alloys.23 Our results agree well with previous experimental findings.24 In addition, the difference of K, G, Y and ν between Li7Sn3, Li5Sn2, Li13Sn5 and Li7Sn2 is minimal.
3.3 Anisotropy
Due to the anisotropy of active materials, micro-cracks can be induced in LIBs during the charging–discharging process. To study the influence of anisotropy on their mechanical properties, we have calculated the elastic anisotropy of Li–Sn alloys by using the universal anisotropy index AU,25 that is| |
 | (13) |
According to Huggins26 and Courtney et al.,27 Li–Sn alloys can be classified into the following three categories based on their Li ion concentration x, viz. (i) the poor lithium phase (x < 2.33), (ii) the medium intercalated-lithium phase (2.33 < x < 3.5) and (iii) the rich lithium phase (3.5 < x < 4.4). As shown in Fig. 2, the elastic anisotropy of the pure metal β-Sn is similar to the poor lithium phases Li2Sn5 and LiSn with the universal anisotropy index AU of 0.44, 0.46 and 0.42, respectively. Li7Sn3 lies in the boundary between the poor lithium and medium intercalated-lithium phases and Li7Sn2 in the boundary between the medium intercalated-lithium and rich lithium phases. Furthermore, both Li7Sn3 and Li7Sn2 exhibit roughly the same universal anisotropy index with the medium intercalated-lithium phases Li5Sn2 and Li13Sn5. This is consistent with the calculated elastic properties (see in Fig. 1), where K, G, Y and ν of these four phases are nearly similar. The full lithiation phase Li17Sn4 trends to be isotropic with AU ≈ 0 since (KV/KR) ≈ 1, which is in agreement with what Ranganathan and Ostoja-Starzewski have suggested.25 A clear anisotropic property can be found for pure metal Li based on the AU value of 7.34, as indicated by former theoretical and experiment results.22,25
 |
| | Fig. 2 Universal anisotropy factor AU at different Li fractions y: (I) the poor lithium phase (x < 2.33), (II) the medium intercalated lithium phase (2.33 < x < 3.5), and (III) the rich lithium phase (3.5 < x < 4.4). | |
3.4 Ductility and brittleness
According to the empirical Pugh formula,28 the ratio G/K can be used to distinguish the ductile and brittle behaviors of intermetallic and metalloid compounds. In the case of G/K > 0.5, a material behaves in a brittle manner, otherwise it is ductile. Here, it is worth noting that, due to the over-simplification, the ductile to brittle transition value of 0.5 is not definitive. However, the G/K ratio during lithiation can describe the change tendency of electrode material properties from ductility to brittleness. It is seen in Fig. 3 that, for pure metal β-Sn and Li, the values of G/K are less than 0.5, and thus they are ductile materials. Obviously, Li–Sn alloys exhibit brittle behaviors because their G/K values are more than 0.5. In addition, Poisson's ratios can also determine the ductile and brittle behaviors. For ductile materials, their Poisson's ratios are generally more than 1/3; but for brittle materials, they are less than 1/3. Thus, we can conclude that Li–Sn alloys are brittle materials because of their low Poisson's ratios (see Fig. 3).
 |
| | Fig. 3 (a) G/K and (b) Poisson's ratio ν versus y = x/(x + 1). | |
In the charging process, electrode materials experience a large-volume deformation with inserting Li ions.29 In addition, electrode material properties may will change from ductility to brittleness. Hence, the brittle active materials are more seriously damaged than ductile ones at the same condition when the Li diffusion-induced stress of external electrode materials transforms from compression to tension.30 In the discharging process, the volume of electrode materials shrinks due to delithiation of Li ions. In the meantime, the average stress upon external electrode materials is tensile, which promotes the propagation of cracks from the external surface to the interior of electrode materials. As the charge–discharge process continues, more cracks appear on the surface and in the interior of electrode materials. This significantly affects the efficiency of active materials and leads to degradation of cycle performance. Thus, it is the ductile–brittle transition of electrode material properties that results in the poor cycle performance and crushing failure of Sn anode materials in the charge–discharge process.
3.5 Electronic structure
To further elucidate the similarity between the mechanical properties of Li7Sn3, Li5Sn2, Li13Sn5 and Li7Sn2, we calculate the electronic structures of Li–Sn alloys. As shown in Fig. 4, there are finite values at the Fermi levels, indicating their metallic conductivities. In addition, the fluctuant N(EF) values near the Fermi level (EF) demonstrate their different electronic conductivity. The partial density of states (PDOS) shows that the Sn 5p states makes the main contribution at EF for most of Li–Sn alloys, but its role is weaken with the increase of Li ion concentration. As shown in Fig. 4(h), in the fully lithiation phase Li17Sn4, the most contribution comes from Li s states due to Li being the dominant constituent atoms.
 |
| | Fig. 4 The calculated total and partial density of states of Li–Sn alloys in the units of states per eV per unit cell and states per eV per atom, respectively. | |
In Li–Sn alloys (except Li17Sn4), the typical feature is the presence of a pseudo-gap (a sharp valley around the Fermi energy). There are two mechanisms for the formation of pseudo-gap in the binary alloys. One is of ionic origin and the other is owing to hybridization effects.23 The pseudo-gap in Li–Sn alloys is believed to be due to covalent hybridization between Sn and Li atoms. Such a strong hybridization gives not only an important mixing between the states of conduction bands but also leads to a separation of the bonding states.31 Thus, Li7Sn3, Li5Sn2, Li13Sn5 and Li7Sn2 show the similar extent of covalent hybridization because of their minor difference of Li contents (∼0.008–0.078), which further results in their similar mechanical properties. It is also see that pseudo-gap appears with the insertion of Li ion, and becomes larger with increasing Li ion, and at high Li ion concentration (Li17Sn4), it disappears. These phenomena correspond to the Sn–Sn covalent bonds that are gradually replaced by the weaker Li–Sn and Li–Li bonds during lithiation. When the Li ion concentration reaches a certain extent, Sn–Sn covalent bonds disappear, and Li–Sn bonds reach the maximum. Then, metallic Li–Li bonds begin to replace Li–Sn bonds with the further increase of Li content. At last, covalent hybridization between Sn and Li atoms disappears. The influence of the change of bonds on the alloys is that the average strength of constituent chemical bonds is weakened, resulting in a weakening tendency of their resistant ability to deformation with the increase of Li concentration. Thus, it is the average weakening bond strength that results in the almost linearly decreased moduli of Li–Sn alloys with increasing Li ion concentration.
4. Conclusions
Based on the density functional theory calculations, we have systematically studied the Li concentration-dependent mechanical properties of Li–Sn alloys. The results show that the bulk, shear and Young's moduli of isotropic LixSn alloys decrease almost linearly with increasing Li concentration, demonstrating that the resistant ability of Li–Sn alloys to deformation is weaken with the increase of Li concentrations. Together with the ductile–brittle transition of electrode materials with the insertion of Li ion, a better understanding has been obtained on the failure of Sn anode materials during the charging–discharging process. It is believed that the results provide a meaningful guide for the improvement of electrode materials of lithium ion batteries.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant nos 11372267 and 11402086), and the National High Technology Research and Development Program of China (863 Program) (Grant no 2013AA032502). Simulations were conducted at iVEC through using advanced computing resources located at iVEC@Murdoch.
References
- N. Alias and A. A. Mohamad, J. Power Sources, 2015, 274, 237–251 CrossRef CAS PubMed.
- H. Liu, Z. Li, Y. Liang, R. Fu and D. Wu, Carbon, 2015, 84, 419–425 CrossRef CAS PubMed.
- F. S. Li, Y. S. Wu, J. Chou, M. Winter and N. L. Wu, Adv. Mater., 2015, 27, 130–137 CrossRef CAS PubMed.
- B. Luo and L. Zhi, Energy Environ. Sci., 2015, 8, 456–477 CAS.
- Z. S. Ma, Y. C. Zhou, J. Liu, D. F. Xue, Q. S. Yang and Y. Pan, Adv. Mech., 2013, 43, 581–599 Search PubMed.
- M. Obrovac and V. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS PubMed.
- J. Wang, F. Fan, Y. Liu, K. L. Jungjohann, S. W. Lee, S. X. Mao, X. Liu and T. Zhu, J. Electrochem. Soc., 2014, 161, F3019–F3024 CrossRef CAS PubMed.
- Y.-T. Cheng and M. W. Verbrugge, J. Power Sources, 2009, 190, 453–460 CrossRef CAS PubMed.
- Y.-T. Cheng and M. W. Verbrugge, J. Appl. Phys., 2008, 104, 083521 CrossRef PubMed.
- J. Wolfenstine, Mater. Lett., 2003, 57, 3983–3986 CrossRef CAS.
- T. Maxisch and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 174112 CrossRef.
- V. B. Shenoy, P. Johari and Y. Qi, J. Power Sources, 2010, 195, 6825–6830 CrossRef CAS PubMed.
- D. H. Chung and W. R. Buessem, J. Appl. Phys., 1968, 39, 2777–2782 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- M. Segall, P. J. Lindan, M. a. Probert, C. Pickard, P. Hasnip, S. Clark and M. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- X. Meng, X. Wen and G. Qin, Comput. Mater. Sci., 2010, 49, S372–S377 CrossRef PubMed.
- B. Narayanan, I. E. Reimanis, E. R. Fuller Jr and C. V. Ciobanu, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 104106 CrossRef.
- K. Panda and K. Ravi Chandran, Comput. Mater. Sci., 2006, 35, 134–150 CrossRef CAS PubMed.
- P. Söderlind and J. E. Klepeis, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 104110 CrossRef.
- W. F. Gale and T. C. Totemeier, Smithells metals reference book, Butterworth-Heinemann, 2003 Search PubMed.
- R. Felice, J. Trivisonno and D. Schuele, Phys. Rev. B: Solid State, 1977, 16, 5173–5184 CrossRef CAS.
- K. Li, H. Xie, J. Liu, Z. Ma, Y. Zhou and D. Xue, Phys. Chem. Chem. Phys., 2013, 15, 17658–17663 RSC.
- J. Chen, S. Bull, S. Roy, H. Mukaibo, H. Nara, T. Momma, T. Osaka and Y. Shacham-Diamand, J. Phys. D: Appl. Phys., 2008, 41, 025302 CrossRef.
- S. Ranganathan and M. Ostoja-Starzewski, Phys. Rev. Lett., 2008, 101, 055504 CrossRef.
- R. A. Huggins, Solid State Ionics, 1998, 113–115, 57–67 CrossRef CAS.
- I. A. Courtney, W. R. McKinnon and J. R. Dahn, J. Electrochem. Soc., 1999, 146, 59–68 CrossRef CAS PubMed.
- S. Pugh, Philos. Mag., 1954, 45, 823–843 CrossRef CAS.
- E. Rejovitzky, C. V. D. Leo and L. Anand, J. Mech. Phys. Solids, 2015, 78, 210–230 CrossRef CAS PubMed.
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, ACS Nano, 2012, 6, 1522–1531 CrossRef CAS PubMed.
- D.-C. Tian and X.-B. Wang, J. Phys.: Condens. Matter, 1992, 4, 8765 CrossRef CAS.
- C. S. Barrett and T. B. Massalski, Structure of metals, McGraw-Hill, New York, 1966 Search PubMed.
- D. Hansen and L. Chang, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 2392–2395 CrossRef CAS.
- W. Muller and H. Schafer, Z. Naturforsch. B: Chem. Sci., 1973, 246–248 Search PubMed.
- W. Müller, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1974, 29, 304–311 Search PubMed.
- U. Frank, W. Müller and H. Schäfer, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1975, 30, 1–5 Search PubMed.
- U. Frank and W. Müller, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1975, 30, 316–322 Search PubMed.
- U. Frank, W. Müller and H. Schäfer, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1975, 30, 6–9 Search PubMed.
- C. Lupu, J.-G. Mao, J. W. Rabalais, A. M. Guloy and J. W. Richardson, Inorg. Chem., 2003, 42, 3765–3771 CrossRef CAS PubMed.
- J. I. Gersten and F. W. Smith, The physics and chemistry of materials, Wiley, New York, 2001 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.