
A robust site-specific Au@SiO2@AgPt nanorod/nanodots superstructure for in situ SERS monitoring of catalytic reactions†
Abstract
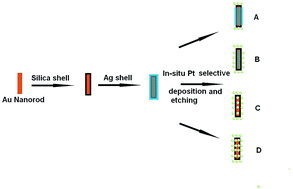
Platinum nanodots have been regiospecifically assembled on a robust core–shell nanorod via an in situ selective deposition and etching protocol. Therefore, different assembled superstructures, including tip coated nanorod/nanodots, tip/edge coated nanorod/nanodots, and core–satellite nanorod/nanodots, have been yielded. In addition, the SERS properties of the assembled superstructures have been investigated, suggesting that the tip coated or tip/edge coated superstructures are superior to the core–satellite superstructures. On the other hand, studies suggest that the catalytic properties of the tip/edge coated and core–satellite superstructure can be better than the tip coated superstructures. Moreover, the tip/edge coated and core–satellite nanorod/nanodots superstructures have been employed as nanoreactors for the real-time SERS monitoring of catalytic reactions.
 Please wait while we load your content...
Please wait while we load your content...

