
Binding to gold nanoclusters alters the hydrogen bonding interactions and electronic properties of canonical and size-expanded DNA base pairs†
Abstract
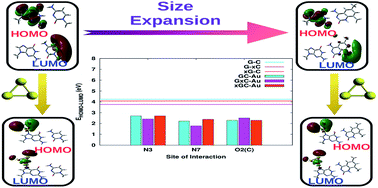
DNA molecules tagged to metal nanoparticles, especially gold nanoparticles (AuNPs), have been shown to have potential applications in the design and fabrication of novel electronic nano-devices, but the binding mechanism between gold nanoparticles and DNA bases and its implications are not completely understood. In this work, a comprehensive study to examine the effect of structural perturbations caused to DNA base pairs in terms of size expansion and adsorption on a gold cluster (Au3) has been carried out using density functional theory. The geometric and electronic features of these complexes provide evidence for the distortion of certain base pairs depending on the binding site of the cluster. This is further substantiated via normal mode, natural bond orbital (NBO) and atoms in molecules (AIM) analyses. The natural population analysis (NPA) and NBO analysis indicate that complexation greatly affects the charge distribution on the base pairs due to charge transfer between the base pair and gold cluster. This charge redistribution may offer the possibility of higher order interactions. Upon complexation, a marked decrease in the HOMO–LUMO gap is observed, which is more profound in cases where size-expanded bases are involved due to the extended π-conjugation of the fused benzene rings. This study demonstrates the possibility of combining structural modifications to DNA base pairs and subsequent binding to gold nanoparticles to modulate and achieve molecular systems with desired optoelectronic properties.
 Please wait while we load your content...
Please wait while we load your content...

