
Metal-free regioselective C-3 nitration of quinoline N-oxides with tert-butyl nitrite†
Jingjing Zhaoab,
Pan Lia,
Chungu Xiaa and
Fuwei Li*a
aState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou, 730000, China. E-mail: fuweili@licp.cas.cn; Fax: +86-931-4968129
bGraduate University of Chinese Academy of Sciences, Beijing, 100049, China
First published on 1st April 2015
Abstract
A direct and eco-friendly nitration methodology to synthesize 3-nitroquinoline N-oxides from quinoline N-oxides using tert-butyl nitrite as both the nitro source and oxidant has been developed. Although this reaction undergoes a free radical process, it exhibits high regioselectivity and can be smoothly scaled up to gram scale.
Aromatic nitro compounds are important and versatile building blocks in synthetic organic chemistry, and their derivatives are widely utilized in various fields such as pharmaceuticals, dyes, explosives, plastics, and materials.1,2 Traditional nitration methods often use an excess of nitric acid or its mixture with sulfuric acid or dinitrogen pentoxide, which then result in limited functional group tolerance under such harsh conditions. Moreover, undesirable by-products and acidic waste are generated by these procedures. Recently, nitro reactions have developed rapidly with the emergence of new nitrating agents such as nitrate,3 nitrite salts4 and tert-butyl nitrite (TBN).5
Recently, direct C–H nitration of olefins and arenes has been developed rapidly. Accordingly, various nitroolefins were synthesized from olefins with different nitrating agents (Scheme 1, Route A).6 Besides, direct C–H nitration of arenes could also be realized through the following two pathways: (1) Pd, Cu or Rh-catalyzed aromatic C–H nitration with the necessary assistance of directing groups (Scheme 1, Route B);7 (2) nitration at the ortho- and para-position of phenol or amines (Scheme 1, Route C).8 Although these nitration methods have gained significant developments, the direct nitration of heterocycles has rarely been reported.
 | ||
| Scheme 1 Direct C–H nitration of olefins and arenes. | ||
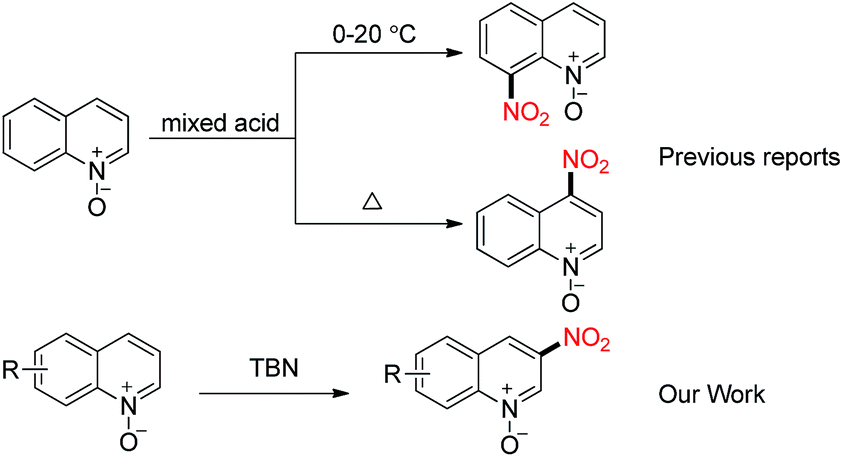
Quinoline N-oxides and quinoline derivatives exist widely in natural products, pharmaceuticals and functional materials. Generally, quinoline N-oxides9 were used as a typical substrate to realize C-2 functionalization via C–H bond activation.10 Meanwhile, the nitration of quinoline N-oxides usually proceed at the 4 or 8-position in the presence of mixed acid (Scheme 2).11,12 Comparing with the above mentioned C2- and C4-functionalization of quinoline N-oxide, the direct C3-functionalization including the nitration has rarely been achieved. Considering that the electron density of C3 position is slightly higher than that of C2 and C4 sites, it is speculated to provide a way to develop selective C3-functionalization method through electrophilic radical coupling strategy. As part of our continuing interest in free radical and the heterocyclic chemistry,13 we herein disclose our investigation on the direct C3 nitration of quinoline N-oxide using TBN as an eco-friendly nitration source under metal free conditions. Although it is known that the selectivity of free-radical transformation is difficult to control, our methodology displays excellent C3 selectivity and can be smoothly scaled up.
 | ||
| Scheme 2 The nitration of quinoline N-oxides at different positions. | ||
Quinoline N-oxide 1a was initially chosen as the model substrate to screen the various reaction parameters (Table 1). The 3-nitroquinoline N-oxide 2a was isolated in 50% yield using NaNO2 as NO2 source with the assistance of Pd(OAc)2 and K2S2O8 in MeCN (entry 1). However, only trace amount of 2a was observed in the absence of Pd(OAc)2 (entry 2). Surprisingly, when TBN was employed as the nitro source and oxidant, the desired 2a was obtained in 77% yield (entry 3). Running the reaction under oxygen or argon atmosphere made the yield decrease to 53% and 66%, respectively (entries 4 and 5). The reaction proceeded smoothly in DCE and toluene (entries 6 and 7). Unfortunately, the reaction did not work in H2O (entry 8). To be noteworthy, 94% yield could be obtained when the loading of TBN was increased to 3.5 equivalent (entry 10). In addition, decreasing the temperature brought a distinct decrease in the yields (entries 11–13), implying such reaction is quite sensitive to temperature variation.
|
||||
|---|---|---|---|---|
| Entry | NO2 source (equiv.) | Solvent | T (°C) | Yield (%) |
| a All reactions were carried out on a 0.3 mmol scale in 3 mL of solvent for 24 h. Isolated yield.b K2S2O8 (2.5 equiv.) was added.c Pd(OAc)2 (10 mmol%) was added.d Under oxygen in pressure tubes.e Under argon in pressure tubes. | ||||
| 1b,c | NaNO2 (2.5) | MeCN | 100 | 50 |
| 2b | NaNO2 (2.5) | MeCN | 100 | Trace |
| 3 | t-BuONO (2.5) | MeCN | 100 | 77 |
| 4d | t-BuONO (2.5) | MeCN | 100 | 53 |
| 5e | t-BuONO (2.5) | MeCN | 100 | 66 |
| 6 | t-BuONO (2.5) | DCE | 100 | 74 |
| 7 | t-BuONO (2.5) | Toluene | 100 | 72 |
| 8 | t-BuONO (2.5) | H2O | 100 | N.R. |
| 9 | t-BuONO (3.0) | MeCN | 100 | 89 |
| 10 | t-BuONO (3.5) | MeCN | 100 | 94 |
| 11 | t-BuONO (3.5) | MeCN | 80 | 74 |
| 12 | t-BuONO (3.5) | MeCN | 50 | 28 |
| 13 | t-BuONO (3.5) | MeCN | 25 | N.R. |

With these satisfactory conditions in hand, we then turned to examining the scope of quinoline N-oxides for this transformation (Table 2). Quinoline N-oxides containing electron-donating groups (OMe, Me, Ph) and electron-withdrawing groups (Br, Cl) at the 5, 6, 7, 8-positions all underwent nitration smoothly at the 3-position. 5-Bromo, 5-methoxy and 5-phenyl quinoline N-oxides gave the desired products (2b–2d) in 60–75% yields, respectively. Likewise, 6-substitued quinoline N-oxides yielded 60–82% products (2e–2i). Meanwhile, 7-methyl quinoline N-oxide afforded 2j in 64% yield. Besides, 8-substituted quinoline N-oxides still gave the desired products (2k and 2l) in moderate yields. Unfortunately, the 2,4-substituted quinoline N-oxides were not suitable for this transformation.14 No products were afforded when isoquinoline N-oxide and pyridine N-oxide were utilized as the substrates. The structure of the nitroquinoline N-oxide 2j was further confirmed by X-ray crystallography (XRD) (CCDC 1031244).


In order to prove the practicality of this approach, a gram-scale synthesis of the 3-nitroquinoline N-oxides 2a (1.36 g, 72% yield) was performed, which suggested that such methodology could also be efficiently scaled up (Scheme 3).
 | ||
| Scheme 3 Gram scale synthesis of 2a. | ||
Furthermore, 3-nitroquinoline N-oxides 2a could be selectively reduced by PCl3 to give the corresponding 3-nitroquinoline 3a in 92% yield. Compound 2a could be hydrogenated to 3-aminoquinoline 3b in 74% yield using Pd2@NAC-800 catalyst15 under 1 atm H2 at room temperature (Scheme 4).
 | ||
| Scheme 4 Transformations of 3-nitroquinoline N-oxide. | ||
In order to deeply understand the mechanism, a series of control experiments were carried out (Scheme 5). The addition of either TEMPO (2,2,6,6-tetramethylpiperidinooxy) or BHT (2,6-di-tert-butyl-4-methylphenol) could stop such nitration reaction (Scheme 5, eqn (a) and (b)). The reaction was also inhibited by 1,1-diphenylethylene, and 1,1-diphenyl-2-nitroethylene was isolated in 55% yield (Scheme 5, eqn (c)).6b These results may suggest that the reaction undergo a free radical process and NO2 radical was involved in present transformation. After the reaction, tertiary butanol was detected by GC-MS, which showed that t-BuONO may generate tBuO radical and NO radical, and the latter could be easily oxidized to NO2 radical via a SET process.16
 | ||
| Scheme 5 Radical trapping experiments. | ||
Based on the above control experiments, a plausible mechanism was proposed as shown in Scheme 6. Initially, TBN generates NO radical, which could be oxidized to NO2 radical via a SET process.5c,16 Then the NO2 radical and quinoline N-oxides generated radical A through an electrophilic radical addition process. Eventually, the desired product was generated via a single-electron oxidation process with the assistance of NO2 or tBuO radical.
 | ||
| Scheme 6 Plausible reaction mechanism. | ||
In summary, we have successfully developed a direct and eco-friendly methodology for the regioselective synthesis of 3-nitroquinoline N-oxides in the absence of any metal catalyst and external oxidant. This transformation is realized due to the higher electron density of C3 position and the electrophilic property of NO2 radical. Furthermore, it is worth noting that such transformation could also be smoothly scaled up. The reactions of free radicals with other coupling partners are under investigation in our laboratory.
Acknowledgements
We gratefully acknowledge the Chinese Academy of Sciences and the National Natural Science Foundation of China (21133011 and 21373246) and the “Strategic Priority Research Program” of the Chinese Academy of Sciences (XDA09030104) for generous financial support.Notes and references
- For an excellent review on aromatic nitro compounds, see: G. Yan and M. Yang, Org. Biomol. Chem., 2013, 11, 2554 CAS.
- (a) N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, 2001 CrossRef; (b) K. Schofield, Aromatic Nitrations, Cambridge University Press, Cambridge, 1980 Search PubMed; (c) H. Feuer and A. T. Nielson, Nitro Compounds: Recent Advances in Synthesis and Chemistry, VCH, Weinheim, 1990 Search PubMed; (d) G. A. Olah, R. Malhotra and S. C. Narang, Nitration: Methods and Mechanisms, VCH, Weinheim, 1989 Search PubMed.
- For selected reports on nitrate salts as the nitro source, see: (a) S. Manna, S. Maity, S. Rana, S. Agasti and D. Maiti, Org. Lett., 2012, 14, 1736 CrossRef CAS PubMed; (b) N. Nowrouzi, A. M. Mehranpour, E. Bashiri and Z. Shayan, Tetrahedron Lett., 2012, 53, 4841 CrossRef CAS PubMed; (c) M. A. Zolfigol, A. Khazaei, A. R. Moosavi-Zare, A. Zare, H. G. Kruger, Z. Asgari, V. Khakyzadeh and M. Kazem-Rostami, J. Org. Chem., 2012, 77, 3640 CrossRef CAS PubMed; (d) J. Jacoway, G. G. K. S. N. Kumar and K. K. Laali, Tetrahedron Lett., 2012, 53, 6782 CrossRef CAS PubMed.
- For selected reports on nitrite salts as the nitro source, see: (a) Y.-M. Li, X.-H. Wei, X.-A. Li and S.-D. Yang, Chem. Commun., 2013, 49, 11701 RSC; (b) B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 12898 CrossRef CAS PubMed; (c) P. J. A. Joseph, S. Priyadarshini, M. L. Kantam and H. Maheswaran, Tetrahedron Lett., 2012, 53, 1511 CrossRef PubMed; (d) G. Yan, L. Zhang and J. Yu, Lett. Org. Chem., 2012, 9, 133 CrossRef CAS; (e) H. J. Yang, Y. Li, M. Jiang, J. M. Wang and H. Fu, Chem.–Eur. J., 2011, 17, 5652 CrossRef CAS PubMed.
- (a) X. Wu, J. Schranck, H. Neumann and M. Beller, Chem. Commun., 2011, 47, 12462 RSC; (b) X. Wu, H. Neumann and M. Beller, Chem. Commun., 2011, 47, 7959 RSC; (c) T. Shen, Y. Yuan and N. Jiao, Chem. Commun., 2014, 50, 554 RSC; (d) S. Wang, C. Shu, T. Wang, J. Yu and G. Yan, Chin. Chem. Lett., 2012, 23, 643 CrossRef CAS PubMed.
- (a) S. Maity, S. Manna, S. Rana, N. Togati, A. Mallick and D. Maiti, J. Am. Chem. Soc., 2013, 135, 3355 CrossRef CAS PubMed; (b) S. Maity, N. Togati, U. Sharma and D. Maiti, Org. Lett., 2013, 15, 3384 CrossRef CAS PubMed; (c) S. Manna, S. Jana, T. Saboo, A. Maji and D. Maiti, Chem. Commun., 2013, 49, 5286 RSC; (d) N. Togati, S. Maity, U. Sharma and D. Maiti, J. Org. Chem., 2013, 78, 5949 CrossRef PubMed; (e) I. Jovel, S. Prateeptongkum, R. Jackstell, N. Vogl, C. Weckbecker and M. Beller, Adv. Synth. Catal., 2008, 350, 2493 CrossRef CAS.
- (a) Y.-F. Liang, X. Li, X. Wang, Y. Yan, P. Feng and N. Jiao, ACS Catal., 2015, 5, 1956 CrossRef CAS; (b) F. Xie, Z. Qi and X. Li, Angew. Chem., Int. Ed., 2013, 52, 11862 CrossRef CAS PubMed; (c) L. Zhang, Z. Liu, H. Li, G. Fang, B.-D. Barry, T. A. Belay, X. Bi and Q. Liu, Org. Lett., 2011, 13, 6536 CrossRef CAS PubMed; (d) Y.-K. Liu, S.-J. Lou, D.-Q. Xu and Z.-Y. Xu, Chem.–Eur. J., 2010, 16, 13590 CrossRef CAS PubMed; (e) D. Katayev, K. F. Pfister, T. Wendling and L. J. Gooßen, Chem.–Eur. J., 2014, 20, 9902 CrossRef CAS PubMed; (f) B. Majhi, D. Kundu, S. Ahammed and B. C. Ranu, Chem.–Eur. J., 2014, 20, 9862 CrossRef CAS PubMed; (g) W. Zhang, S. Lou, Y. Liu and Z. Xu, J. Org. Chem., 2013, 78, 5932 CrossRef CAS PubMed; (h) P. Sadhu, S. K. Alla and T. Punniyamurthy, J. Org. Chem., 2014, 79, 8541 CrossRef PubMed.
- (a) Y.-X. Li, L.-H. Li, Y.-F. Yang, H.-L. Hua, X.-B. Yan, L.-B. Zhao, J.-B. Zhang, F.-J. Ji and Y.-M. Liang, Chem. Commun., 2014, 50, 9936 RSC; (b) D. Koley, O. C. Colón and S. N. Savinov, Org. Lett., 2009, 11, 4172 CrossRef CAS PubMed; (c) B. Kilpatrick, M. Heller and S. Arns, Chem. Commun., 2013, 49, 514 RSC; (d) A. K. Bose, S. N. Ganguly, M. S. Manhas, S. Rao, J. Speck, U. Pekelny and E. Pombo-Villars, Tetrahedron Lett., 2006, 47, 1885 CrossRef CAS PubMed.
- The quinoline N-oxides were easily prepared from quinoline with m-CPBA, see: G. Li, C. Jia and K. Sun, Org. Lett., 2013, 15, 5198 CrossRef CAS PubMed.
- (a) G. Yan and M. Yang, Adv. Synth. Catal., 2014, 356, 2375 CrossRef CAS; (b) J. Wu, X. Cui, L. Chen, G. Jiang and Y. Wu, J. Am. Chem. Soc., 2009, 131, 13888 CrossRef CAS PubMed; (c) X. Chen, C. Zhu, X. Cui and Y. Wu, Chem. Commun., 2013, 49, 6900 RSC; (d) Z. Wu, C. Pi, X. Cui, J. Bai and Y. Wu, Adv. Synth. Catal., 2013, 355, 1971 CrossRef CAS; (e) G. Li, C. Jia and K. Sun, Org. Lett., 2013, 15, 5198 CrossRef CAS PubMed; (f) S. H. Cho, S. J. Hwang and S. Chang, J. Am. Chem. Soc., 2008, 130, 9254 CrossRef CAS PubMed; (g) X. Gong, G.-Y. Song, H. Zhang and X.-W. Li, Org. Lett., 2011, 13, 1766 CrossRef CAS PubMed; (h) Y. Shen, J. Chen, M. Liu, J. Ding, W. Gao, X. Huang and H. Wu, Chem. Commun., 2014, 50, 4292 RSC; (i) Z. Wu, H. Song, X. Cui, C. Pi, W. Du and Y. Wu, Org. Lett., 2013, 15, 1270 CrossRef CAS PubMed.
- (a) T. Kaiya, N. Shirai and Y. Kawazoe, Chem. Pharm. Bull., 1986, 34, 881 CrossRef CAS; (b) L. H. Heitman, A. Goblyos, A. M. Zweemer, R. Bakker, T. Mulder-Krieger, J. P. D. Veldhoven, H. Veies, J. Brussee and A. P. IJzerman, J. Med. Chem., 2009, 52, 926 CrossRef CAS PubMed.
- Only one example of 3-nitroquinoline N-oxides was reported, using stoichiometric AgNO3 as the nitrating agent, but only in 33% yield, see: K. S. Sharma, S. Kumari and R. P. Singh, Synthesis, 1981, 316 CrossRef CAS.
- (a) J. Zhao, P. Li, C. Xia and F. Li, Chem. Commun., 2014, 50, 4751 RSC; (b) P. Li, J. Zhao, C. Xia and F. Li, Org. Lett., 2014, 16, 5992 CrossRef CAS PubMed; (c) P. Li, J. Zhao, R. Lang, C. Xia and F. Li, Tetrahedron Lett., 2014, 55, 390 CrossRef CAS PubMed; (d) Q. Xing, P. Li, H. Lv, R. Lang, C. Xia and F. Li, Chem. Commun., 2014, 50, 12181 RSC.
- Possibly owing to the steric effect of 2 or 4-position, quinoline N-oxide derivatives (such as 2-Cl, 2-Me, 4-Me, 4-NO2) were not suitable for this transformation.
- The synthesis of Pd2@NAC-800, see: Z. Li, J. Li, J. Liu, Z. Zhao, C. Xia and F. Li, ChemCatChem, 2014, 6, 1333 CAS.
- The NO radical was easily oxidized to NO2 radical in the presence of trace amount of O2 and H2O, see: (a) T. Taniguchi, A. Yajima and H. Ishibashi, Adv. Synth. Catal., 2011, 353, 2643 CrossRef CAS; (b) I. Jovel, S. Prateeptongkum, R. Jackstell, N. Vogl, C. Weckbecker and M. Beller, Adv. Synth. Catal., 2008, 350, 2493 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1031244. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra04632g |
| This journal is © The Royal Society of Chemistry 2015 |