
ESR/UV-Vis-NIR spectroelectrochemical study and electrochromic contrast enhancement of a polythiophene derivative bearing a pendant viologen†
Bhushan Gadgil*ab,
Pia Damlina,
Evgenia Dmitrievac,
Timo Ääritaloa and
Carita Kvarnström*a
aTurku University Centre for Materials and Surfaces (MATSURF), Laboratory of Materials Chemistry and Chemical Analysis, University of Turku, FI-20014, Finland. E-mail: bhushan.gadgil@utu.fi; gadgil.bhush@gmail.com; carkva@utu.fi; Fax: +358 2 333 6700; Fax: +358 2 333 6700; Tel: +358 2 333 6868 Tel: +358 2 333 6729
bUniversity of Turku Graduate School (UTUGS), FI-20014, Turku, Finland
cCenter of Spectroelectrochemistry, IFW Dresden, Helmholtzstrasse 20, D-01069 Dresden, Germany
First published on 7th April 2015
Abstract
The importance of viologens in the field of electrochromic materials is well recognized due to their intensely colored radical cation formation. In this study, 1-[6-[(4-methyl-3-thienyl)oxy]hexyl]-4,4′-bipyridium hexafluorophosphate (Th-V) was synthesized and electropolymerized in a solvent mixture comprising water and acetonitrile (v/v; 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) with 0.1 M lithium perchlorate (LiClO4) as an electrolyte salt, yielding a viologen bearing polythiophene (PTh-V) film on an electrode surface. The resulting polymer shows electrochemical activity from both the redox active viologen and the conjugated polythiophene moieties. The redox behavior of the polymer was studied by multi in situ spectroelectrochemical technique by means of simultaneous recording of electron spin resonance and UV-Vis-near infrared (ESR/UV-Vis-NIR) spectra. The results indicate that only polaron charge carriers are created during both n- and p-doping of the PTh-V film. The polymer film shows enhanced electrochromic contrast due to the introduction of a pendant viologen group into the thiophene unit. The film switched reversibly between dark violet (at −0.6 V) and almost transparent (at 1.0 V) showing good optical contrast with a coloration efficiency of ca. 305 cm2 C−1 at 610 nm. The switching transmittance kinetics demonstrate fast response times to attain a bleached state and excellent operational stability with repeatable voltage switching between colored/bleached states for 1000 cycles. The polythiophene backbone was found to strengthen the thermal stability of the conjugated PTh-V redox polymer. The excellent optical contrast with sharp color changes and high color efficiency combined with adequate thermal behavior suggests the potential of PTh-V in the electrochromic device (ECD) application.
:50) with 0.1 M lithium perchlorate (LiClO4) as an electrolyte salt, yielding a viologen bearing polythiophene (PTh-V) film on an electrode surface. The resulting polymer shows electrochemical activity from both the redox active viologen and the conjugated polythiophene moieties. The redox behavior of the polymer was studied by multi in situ spectroelectrochemical technique by means of simultaneous recording of electron spin resonance and UV-Vis-near infrared (ESR/UV-Vis-NIR) spectra. The results indicate that only polaron charge carriers are created during both n- and p-doping of the PTh-V film. The polymer film shows enhanced electrochromic contrast due to the introduction of a pendant viologen group into the thiophene unit. The film switched reversibly between dark violet (at −0.6 V) and almost transparent (at 1.0 V) showing good optical contrast with a coloration efficiency of ca. 305 cm2 C−1 at 610 nm. The switching transmittance kinetics demonstrate fast response times to attain a bleached state and excellent operational stability with repeatable voltage switching between colored/bleached states for 1000 cycles. The polythiophene backbone was found to strengthen the thermal stability of the conjugated PTh-V redox polymer. The excellent optical contrast with sharp color changes and high color efficiency combined with adequate thermal behavior suggests the potential of PTh-V in the electrochromic device (ECD) application.
Introduction
Electrochromic (EC) materials possess a reversible redox behavior by accepting or donating electrons under an applied DC voltage, simultaneously undergoing optical contrast, switching from one colored state to another.1 A number of different organic and inorganic materials have been tested for improving dynamic and static EC properties such as fast switching time, high optical contrast in the visible region, large coloration efficiency, broad range of colors and enhancing EC contrast.2,3 In the last decade, conducting polymers have gained much attention in the EC field as they are inexpensive, possesses long term stability and offer multiple colors. Polythiophene (PTh) is one of the widely studied conjugated polymers due to its diverse electrochemical properties and strong electrochromism.4 However, the processibilty of pristine PTh seems to be a great problem due to the availability of more than one polymerization site or irreversible side reactions at 3- or 4- positions of the Th ring.5 In order to overcome this problem, researchers have synthesized a thiophene (Th) oligomer which is functionalized with electron-donating groups such as alkyl or alkoxy groups.6,7 Such groups not only protect the 3- and 4- positions from side reactions but also lower the oxidation potential of the monomer.8,9 These aspects have been considered when synthesizing the monomer for this work.Recently, considerable effort has been made to modify the electronic properties of conducting polymers mainly by attaching specific functional groups to the polymeric backbone, the so called third generation of polymers.10–12 Keeping in mind the benefits of conducting polymers, we anticipated that the contrast would be significantly enhanced if one could incorporate an additional EC element into the side chains of PTh. We have selected viologen (V) as such an element owing to its highly stable electrochemical redox reaction leading to cation radical formation with corresponding sharp color changes.13 A convenient way to deposit a conjugated or redox polymer at the electrode surface is by electrochemical polymerization of soluble precursors in a suitable electrolyte. In our previous studies, we attempted to electropolymerize a thiophene monomer bearing a cross-linkable cyanopyridine moiety.14 The aim was to obtain an insoluble polythiophene derivative film containing viologen side groups. However, the film was soluble in the electrolyte possibly due to the bulkiness of the monomer structure. In order to avoid this, we employed an ionic liquid as an electrolyte instead of conventional solvents and successfully obtained a polythiophene film cross-linked with viologens.15 However, the electroactivity of the thiophene moiety was still not to our satisfaction. We therefore synthesized an electropolymerizable 3,4-disubstituted thiophene monomer chemically functionalized with a pendant redox active viologen (Th-V) connected to the side chain at the 3-position of Th. Its electropolymerization thought to yield an insoluble conducting polythiophene film bearing pendant viologen.16
Thorough characterization of organic molecules is difficult by traditional methods due to the complexity of their structures. Moreover, when it comes to the detailed information on the nature of charge carriers, e.g. magnetic or optical features within the molecules, conventional electrochemical methods are insufficient for a concise characterization. In recent years, in situ spectroelectrochemistry has emerged as a useful tool over the traditional electrochemical methods.17,18 Multi-spectroelectrochemistry comprises an electrochemical setup coupled to magnetic and/or optical instrumentation which determines the corresponding information from the organic conjugated materials. Upon electrochemical doping, new electronic states are developed due to the creation of charge carriers in the π-conjugated backbone. This leads to corresponding optical transitions in the UV-Vis-NIR region. Some charge carriers are paramagnetic in nature and can be detected by ESR spectroscopy. Because of both optical and magnetic characteristics in the conjugated organic materials like PTh-V, the combination of ESR and UV-Vis-NIR spectroelectrochemistry is helpful in the elucidation of the doping mechanism as well as to systematically characterize the nature of charge carriers involved during the electrochemical doping/dedoping processes.19,20
In this work, we report the electrochemical polymerization of Th-V monomer in a water–acetonitrile mixture (v/v; 50:50) containing LiClO4 as an electrolyte salt. The electropolymerization gives an insoluble electroactive PTh-V film deposited at the electrode surface. The electrochemical response of the film confirms involvement of both PTh and V species in the polymeric material. In situ ESR/UV-Vis-NIR spectroelectrochemistry is used to determine the doping induced charge carriers created during electrochemical p- and n-doping of PTh-V. The addition of V into the PTh backbone seems to remarkably increase the overall EC contrast of the resultant PTh-V material. Electrochromic switching kinetics showed effective transitions between the bleached and darkened state with abrupt color changes. TGA studies show the adequate thermal stability of the redox polymeric material.
Experimental
Synthesis of N,N′-bis{[6-(4-methyl-3-thienyl)oxy]hexyl}-4,4′-bipyridinium dibromide (Th-V)
3-(6-Bromohexyloxy)-4-methylthiophene (2.61 g; 9.4 mmol) and 4,4′-bipyridine (0.49 g; 3.1 mmol) were dissolved in acetonitrile (5 mL) in a closed cylinder. The cylinder was then placed in an oven at 120 °C for 24 h. A yellow precipitate was formed. After cooling, 40 mL of diethyl ether was added and the crude product was collected by filtration. The crude material was dissolved in methanol and treated with activated carbon, after filtration and evaporation under reduced pressure, 1.7 g of pure material was obtained (Scheme 1). | ||
| Scheme 1 The chemical structure of monomer Th-V. | ||
1H NMR (D2O): 9.16 (4H, d), 8.54 (4H, d), 6.93 (2H, s), 6.43 (2H, s), 4.60 (4H, t), 3.83 (4H, t), 1.98 (4H, m), 1.93 (3H, s), 1.67 (4H, m), 1.40 (8H, m).
13C NMR: 156, 150, 146, 129, 127, 120, 97, 70, 61, 30, 28, 25, 24, 12.
Electrochemistry
The electrochemical polymerization of Th-V and all other electrochemical experiments were performed in a conventional three-electrode one-compartment set-up using a CV technique operated by an Autolab (PGSTAT101) work station under computer control. In the CV experiments, a Au electrode (deposition area 1 cm2) was used as the working electrode and a platinum wire as the counter electrode. A Ag wire coated with AgCl, used as a pseudo-reference electrode, was calibrated using 1 mM ferrocene (Fe/Fe+) in the supporting electrolyte solution (E1/2 (Fe/Fe+) = 0.45 V) before all of the experiments. All following potentials mentioned are vs. the Ag/AgCl electrode. All electrochemical characterizations and CV experiments of PTh-V were performed using a mixture of electrolyte made by 0.1 mole L−1 solution of lithium perchlorate (LiClO4) in the two solvents, viz. deionized water and acetonitrile (v/v; 50:50). The PTh-V film was electrosynthesized by potential cycling from −1.2 to 1.5 V using 30 cycles at a scan rate of 50 mV s−1. The mixed electrolyte solution was deaerated by a dry nitrogen stream for 15 min before each experiment to ensure an oxygen free environment. After each electropolymerization experiment, the polymer film was washed repeatedly with copious amounts of deionized water in order to remove the electrolyte, monomer or oligomers. The deposited films were then placed in a monomer-free electrolyte for CV measurements of the PTh-V film-coated electrode.
Spectroelectrochemistry
In situ ESR/UV-Vis-NIR spectroelectrochemical experiments were performed in an optical ESR cavity (ER 4104OR, Bruker Germany). ESR spectra were recorded using an EMX Micro X-band CW spectrometer (Bruker, Germany). UV-Vis-NIR spectra were recorded using an Avantes spectrometer AvaSpec-2048x14-USB2 with a CCD detector and AvaSpec-NIR 256-2.2 equipped with an InGaAs detector applying the AvaSoft 7.5 software. Both the ESR and the UV-Vis-NIR spectrometer were connected to a HEKA potentiostat PG 390. Triggering was performed by the software package PotMaster v2x40 (HEKA Electronic, Germany). The spectroelectrochemical flat cell with a three-electrode arrangement consisting of a laminated Au mesh as a working electrode, a Pt wire as a counter electrode and a Ag wire coated with AgCl as a pseudo-reference electrode was used. A Au-microgrid (1024 meshes per cm2) was positioned between two pieces of chemically resistant polyester based lamination foils (DocuSeal, USA) to obtain the small well defined electrochemically active surface with an insulated electrical contact, with circular holes giving 0.1 cm2 of free active electrode surface.Characterization
Thermogravimetric analysis (TGA) was performed using a TA Instruments SDT 2960 simultaneous DTA-TGA thermoanalyzer. TGA analysis of the V film was performed in the temperature range of 0–600 °C. The solid state PTh-V material was subjected to thermal degradation in the temperature range between 0 and 1000 °C. The heating rate was 20 °C min−1 and the analysis was carried out under a nitrogen atmosphere. The EC switching kinetic study was performed using a chronoamperometry technique by switching of the potential between 1.0 and −0.6 V at a rate of 10 s. The corresponding optical changes were recorded using an Agilent Cary 60 UV-Vis spectrophotometer. Impedance measurements were performed using an IviumStat potentiostat. EIS studies were made at different dc potentials (from 0 to −1.0 V at the cathodic side and 0 to 1.2 V at the anodic side, ΔEdc = 0.1 V) in the frequency range of 100 kHz–0.1 Hz by using an ac excitation amplitude of 10 mV.Results and discussion
Electropolymerization and CV analysis
The electrochemical polymerization of Th-V (0.02 mol L−1) was performed by cyclic voltammetry (CV), and the resulting CVs are shown in Fig. 1. Since viologen electrochemistry is well studied in aqueous electrolyte while that of conjugated thiophene is well studied in organic media, we employed a mixed solvent strategy comprising water and acetonitrile (v/v; 50:50) as electrolyte with LiClO4 as the supporting salt in order to activate both Th as well as V in their respective electrochemical states. The electrode potential was initially swept to −1.2 V to monitor the electroactivity of the viologen species, and further continued in the anodic direction until 1.5 V to oxidize the Th monomer. From the second sweep on, a well-defined redox response of the PTh moiety appeared and increased successively at ca. 1.0 V upon following scanning. At cathodic potentials, a redox couple at ca. −0.4 V and a broad one at −0.75 V can be observed at potentials identical to that of the viologen unit. Interestingly, an additional peak was observed at ca. −0.2 V which increased with consecutive scanning. After electropolymerization, an obscure violet layer can be seen on the Au electrode surface due to highly cross-linked viologen moieties within the polymer. When compared to the previous works on similar structures,21,22 the electroactivity of the PTh-V film was found to be remarkably stable on both sides and could withstand 30 sweep cycles for electropolymerization.
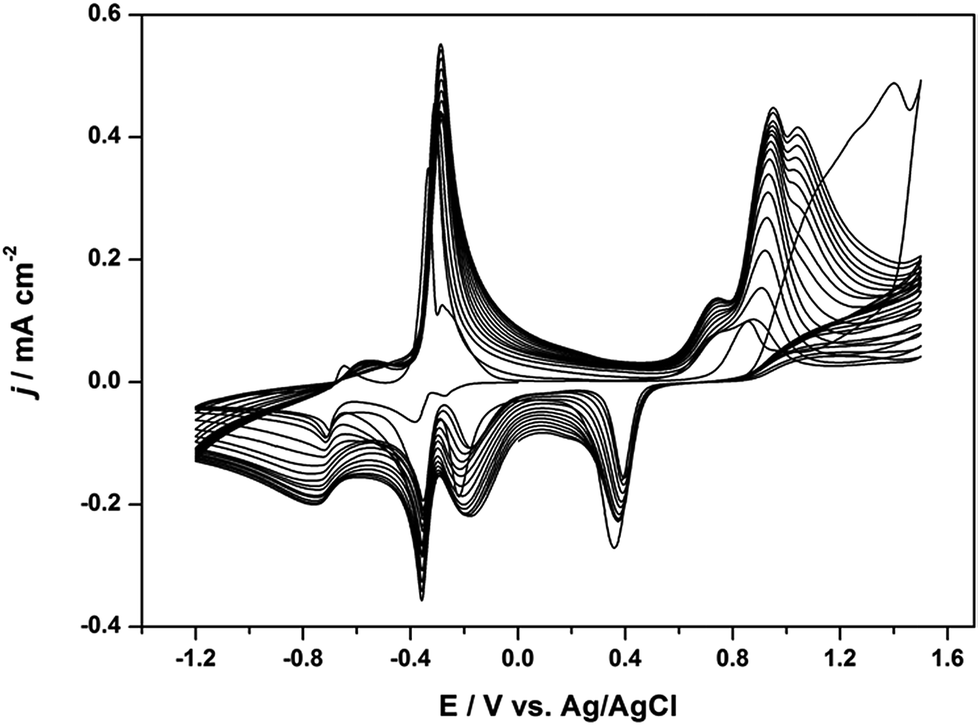 | ||
| Fig. 1 Cyclic voltammograms (CVs) for the electropolymerization of 0.02 mol L−1 Th-V on a Au electrode in a mixture of solvents containing water and acetonitrile (v/v; 50:50) using 0.1 M LiClO4 as the electrolyte salt. Scan rate: 50 mV s−1. | ||
The electroactivity of the deposited polymer, PTh-V, at the Au electrode surface was studied by the CV technique in monomer free electrolyte. As shown in Fig. 2, in the cathodic range, the reduction peak observed at −0.4 V is assigned to the formation of a viologen radical cation which is followed by a very intense oxidation peak at −0.35 in the reverse scan (V2+/V˙+). At more negative potentials, a second broad redox couple reduced at −0.76 V associated with its oxidized counterpart at −0.64 V can be assigned to the neutral viologen species formation (V˙+/V0). A pre-peak was observed at −0.2 V before the radical cation formation was consistent. In the anodic range, an oxidation doublet at 0.8 and 1.0 V can be found which corresponds to the PTh+ moiety along with a reduction peak at 0.4 V.
 | ||
| Fig. 2 Corresponding CV response of PTh-V in water–acetonitrile mixed electrolyte containing 0.1 M LiClO4. Scan rate: 50 mVs−1. | ||
In situ ESR/UV-Vis-NIR spectroelectrochemistry
 | ||
| Fig. 3 In situ (a and c) ESR and (b and d) UV-Vis-NIR spectra of PTh-V film measured during cathodic scan from 0 to −0.6 V and during anodic sweep from 0 and 1.2 V respectively. | ||
Fig. 3b shows the in situ UV-Vis-NIR spectra of the cathodically charged/discharged PTh-V film obtained simultaneously during in situ ESR measurements. At the beginning of the potential scan, no substantial changes are observed in the UV-Vis-NIR spectrum. When the negative potential is increased to −0.4 V the spectra are dominated by three absorption bands. The first band has its maximum at ca. 400 nm while the second and third are broad bands at ca. 550 and 900 nm respectively. These bands are identical with those of the pristine viologen molecule and assigned to the viologen radical formation.24 All three absorptions increase in intensity with increasing applied potential and the band with maxima at ca. 550 nm becomes less broad compared to the one at ca. 900 nm due to more radicals acquired in the film resulting in higher visible absorption. During discharging of the PTh-V film, the three absorption bands decrease gradually until −0.4 V and thereafter, reduce substantially. The absorption spectra thus have a similar potential dependence to the ESR spectra (Fig. S2†) suggesting that both techniques correlate well and that the viologen cation radical is strongly colored, highly stable and paramagnetic in nature.
The in situ UV-Vis-NIR spectra obtained during p-doping of PTh-V are presented in Fig. 3d. As depicted, the changes in absorption intensity correlate well with the ESR signal. The absorbance intensity remains constant until the potential reaches 0.8 V, where two absorption bands start to grow at around 700 nm and 1400 nm. Both bands increase with further increase in potential. Unlike common conducting polymers where further oxidative charging results in polaron pairs or bipolarons,26 no such charge carriers could be detected in the PTh-V film. This might be due to steric hindrance in the precursor structure which after electropolymerization leads to short PTh chains. The only charge carrier formed during the p-doping process would therefore be positive polarons. During dedoping, the intensity decreases gradually and reaches its initial value at the end of the reaction course.
Thus, the in situ ESR/UV-Vis-NIR study shows that polarons were the sole doping induced charge carriers introduced during cathodic and anodic charging of the PTh-V film. Moreover, the polymer structure might contain short chains of oligomers or dimers, due to that bipolaron or polaron pairs could not be observed during PTh doping.
Absorbance spectra and coloration efficiency (CE)
The in situ UV-Vis spectral studies were carried out to observe the absorption changes in PTh-V at different oxidation states and the spectra are shown in Fig. 4. The film thickness was calculated to be ca. 150 nm. In the anodic sweep, the absorption intensity remains constant during applied potentials between 0 and 0.6 V. From 0.8 V onwards, the absorbance at 700 nm starts to grow and the yellowish polymer turned to transparent light blue due to oxidation of PTh-V2+ to PTh+-V2+. During the cathodic sweep, the absorbance at ca. 380, 550 and 900 nm increases suddenly at −0.4 V due to PTh0-V˙+ formation giving a dark violet colored film. As the potential further increases, the absorbance grows until reaching the potential of −0.6 V. At this potential, all radical cation species are consumed and further increase in the applied voltage results in a second viologen redox state PTh0-V0 and a decrease in absorbance at 550 and 900 nm. | ||
| Fig. 4 Static spectroelectrochemistry of PTh-V on ITO glass at different applied potentials. | ||
With further increase in applied voltage, the absorbance at 380 nm shifts to ca. 400 nm and is growing even at −1.2 V due to the radical and the neutral forms of viologen absorbing at the close laying wavelengths. Among these oxidation states, EC contrast was explored in the visible region between PTh+-V2+ and PTh0-V˙+ where the PTh+-V2+ is transparent blue while PTh0-V˙+ is deep violet colored. These potential-dependent optical changes suggest that the EC contrast of PTh was improved by incorporation of the pendant viologen side group. Therefore, an EC response can be described according to the redox reactions operating in the PTh-V film, with a bleached (transparent blue) state at ca. 1.0 V due to oxidized PTh species while the colored state (deep violet) at −0.6 V contributed by the reduced V moieties and can be expressed as:
 | (1) |
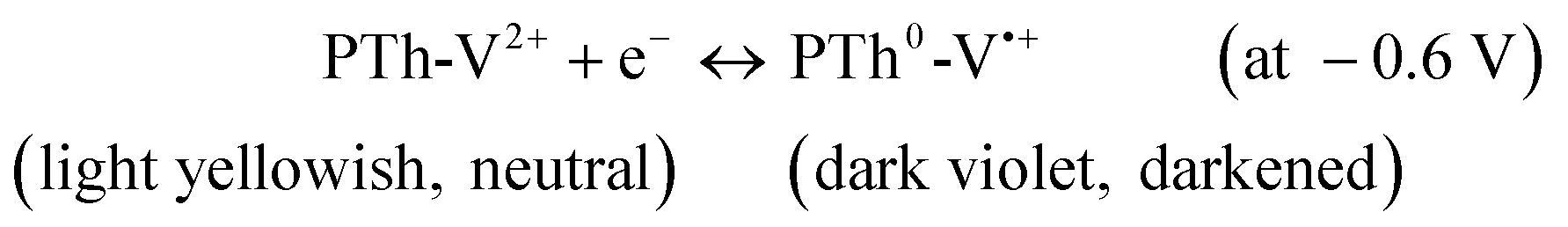 | (2) |
The overall bleaching–darkening transition for a PTh-V film between 1.0 V and −0.6 V can thus be written as:
 | (3) |
In order to characterize the electrochromic response of the PTh-V films, an intermediate wavelength of 610 nm is selected where the contributions from both PTh and V species have been considered. Moreover, in the most darkened state, a tiny broad band at ca. 610 nm was observed in the absorption spectra, most probably due to contributions from both PTh and V. Therefore, 610 nm was chosen as the monochromatic wavelength for measuring the electrochromic properties of the film.
Fig. 5 shows the relationship between the optical density change (ΔOD) at 610 nm and the charge density (Q) for the PTh-V film. Coloration efficiency (CE or η) at a certain monochromatic wavelength can then be calculated from the slope of a straight line derived from the plot of ΔOD vs. Q.27 The reaction charge density was obtained by integrating the i–t curve. The reference state was set at 1.0 V and a series of voltages were applied with a decrement of 0.1 V till −0.6 V with each potential pulse lasting for 60 s to reach equilibrium conditions. The CE for the PTh-V system was calculated to be 305 cm2 C−1 at 610 nm. Our η value is substantially higher than the one reported for poly (butyl viologens) as 163 cm2 C−1 (ref. 28) and almost matches with the value of 342 cm2 C−1 recently reported for electrochromic heptyl viologen.28 An ECD composed of an ionic liquid based gel polymer electrolyte with a functionalized viologen derivative reported a CE of up to 725 cm2 C−1.29 A heptyl viologen composite with In/Sn oxide nanoparticles showed a η value of 912 cm2 C−1.30
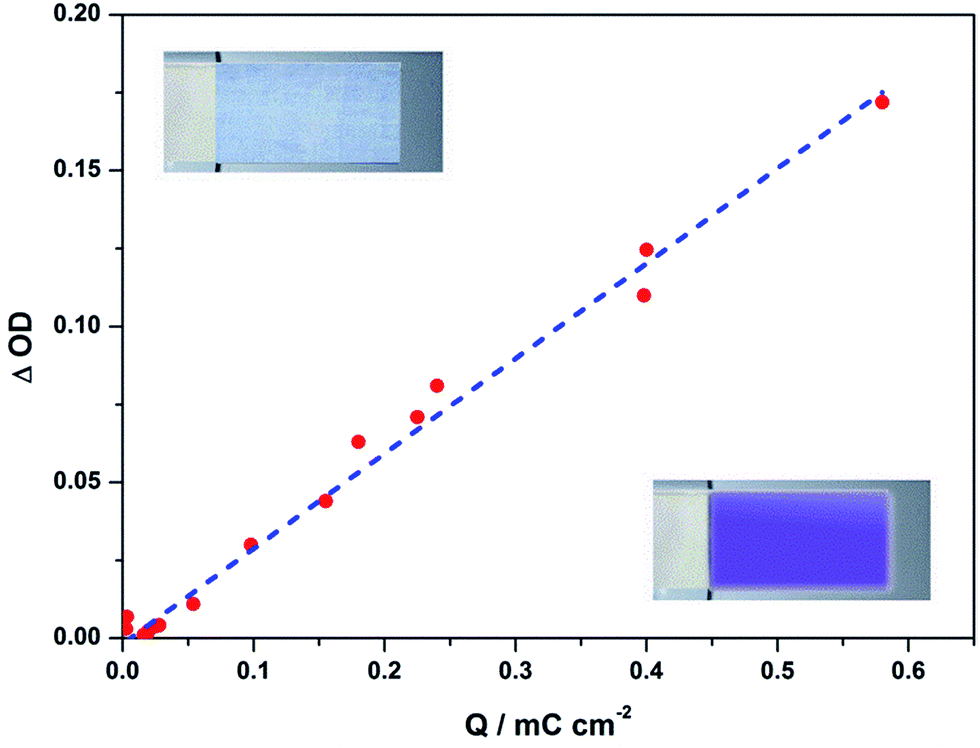 | ||
| Fig. 5 Optical density change (ΔOD) of the PTh-V film at 610 nm as function of charge density (Q) at different applied potentials ranging from 1.0 to −0.6 V. | ||
Electrochromic (EC) switching kinetics
For ECD applications, a polymer material under study should rapidly switch between its reduced and oxidized state with striking color changes.29,30 The color-bleach characteristics of the PTh-V film were recorded at a wavelength (λmax) of 610 nm, with a step time of 10 s and switching the voltage between −0.6 and 1.0 V. Fig. 6a shows optical transmittance kinetic curves during alternate dark and bleach states. Corresponding E–t curves during the switching experiments are shown in Fig. 6b. The %T for the PTh-V film can reach up to 59% for the dark state while for the bleached state it was as low as 20%. The Δ%T was found to be ca. 39% at 610 nm indicating suitability of the PTh-V film in the application of ECDs.31 Coloration and bleaching times, defined as the time required for the EC film to reach 90% transmittance change, were calculated to be 7 s and 5 s, respectively (Fig. 6c). No significant degradation of the film was noticed after 1000 cycles with a step interval of 10 s, as shown in Fig. 6d. | ||
| Fig. 6 (a) Optical transmittance kinetic curves for the PTh-V film measured at 610 nm by switching the voltage from 1.0 to −0.6 V with an interval of 10 s, (b) corresponding I–t curves obtained during optical switching test, (c) switching response time of the PTh-V film and d) cycling stability test for electrochromic PTh-V at bleached (1.0 V) and colored (−0.6 V) states. | ||
A noticeable delay in the EC response can be clearly observed when switching the potentials between the two states. When the applied potential was switched from 1.0 V to −0.6 V, the absorbance and current showed a two-step response, with initial fast changes followed by a slower one. The initial fast change is mainly attributed to the reduction of the conjugated PTh+ moiety to insulated PTh0, complicating the electron transfer from the conducting polymeric phase to the viologen units. This can be seen from the current value during reduction that does not drop to zero, instead decays slowly suggesting that V˙+ is still undergoing reduction. When the applied potential was reversed from −0.6 to 1.0 V, the response was much faster. More than 50% transmittance change was taking place within a second. This might be due to two processes being involved at this stage; one is the oxidation of V˙+ to V2+ while the other one alters insulated PTh into its conducting form.16 Moreover, when PTh gets oxidized to PTh+, it facilitates the oxidation of reduced viologen, making the process faster at 1.0 V. These results are well supported by the electrochemical impedance spectroscopy (EIS) measurements (Fig. 7). The Nyquist plot during V doping shows a semicircle over the entire frequency region, suggesting highly resistive charge transfer behavior of viologen species at the film–electrolyte interface mainly due to the non-conducting V˙+ and insulated PTh. When the polymer film was doped at the anodic direction, the resistive properties of the film get lowered and the plot comprises a line with a slope of 45°, indicating low charge transfer resistance and better conductivity due to the p-doped PTh backbone. The results verified that the conducting properties of the redox V film are improved when combined with conjugated PTh.
 | ||
| Fig. 7 Nyquist plot of the PTh-V film at the cathodic side (V) and at the anodic side (PTh) in the frequency range of 100 kHz to 0.1 Hz. | ||
Table 1 summarizes the electrochemical and electrochromic properties of the PTh-V film. The electrochemical data contains onset oxidation potential (Eoxonset), maximum absorption wavelength (λmax), low energy absorption edges (λonset), HOMO and LUMO energy levels and the optical band (Eg, OPT) values. The electrochromic parameters include monochromatic wavelength used for electrochromic measurements ((λmax), optical transmittance at reduced (Tred) and oxidized states (Tox), change in transmittance (ΔT), response times and coloration efficiency for EC PTh-V.
| Electrochemical data | Electrochromic parameters | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Eoxonset | Eredonset | λonset (nm) | Eg, OPTa (eV) | HOMOb (eV) | LUMOc (eV) | λmax (nm) | Tred | Tox | ΔT | Response time (s) | Coloration efficiency (cm2 C−1) | |
| Oxidation | Reduction | |||||||||||
| a Data were calculated by the equation: Eg = 1240/ λonset.b The HOMO energy level was calculated from CV data using equation HOMO = Eox + 4.4.26c Estimated using empirical equation LUMO = Ered + 4.4. | ||||||||||||
| 0.86 | 0.31 | 750 | 1.65 | 5.26 | 4.09 | 610 | 20 | 58 | 38 | 5 | 7 | 305 |
Thermal stability
Thermal degradation of EC polymers is very important for their potential applications in ECDs. A TGA plot of the electrodeposited V and PTh-V film in the temperature range of 0–1000 °C under a nitrogen atmosphere is shown in Fig. 8. The plot demonstrates the weight loss of the sample as a function of temperature. For the cathodically electrodeposited V film, the initial 10% weight loss comes from adsorbed moisture up to 140 °C, followed by a steady curve until 240 °C. After this temperature, the material degrades quickly and complete degradation occurred at 320 °C. This suggests fast weight loss and low degradation temperature for viologen deposition. For the PTh-V film, a plateau was observed with a small weight loss (3–5%) in the initial phase, due to the evaporation of adsorbed moisture from the PTh-V film until 370 °C, where the initial start of decomposition could be observed. The decomposition of PTh-V polymer at relatively high temperature indicates its suitability for real ECD applications. Further increase in the temperature causes up to 45% weight loss at 500 °C due to major structural decomposition of PTh-V. At 1000 °C, almost 75% of the weight loss was achieved. Normally conducting polymers like polythiophene were reported to have quite high thermal stability due to their rigid polymeric backbone.32 Unlike the low thermal degradation of viologen materials, the incorporation of a conjugated PTh backbone has reasonably improved the thermal properties of PTh-V. | ||
| Fig. 8 TGA plot of cathodically deposited viologen (V) and PTh-V at a heating rate of 20 °C min−1 under a nitrogen atmosphere. | ||
Conclusions
This article describes the novel synthesis and electrochemical properties of a redox polymer, containing a conjugated polythiophene backbone and viologen side group (PTh-V), derived from electropolymerization of thiophene derivative functionalized with viologen (Th-V). Fairly stable films of PTh-V were obtained from a water–acetonitrile mixture containing LiClO4 as an electrolyte salt. The CV response of the films showed contributions from both the redox active viologen moiety and the polythiophene backbone. Doping induced charge carriers determined by in situ ESR/UV-Vis-NIR spectroelectrochemistry were found to be exclusively polarons during both viologen and polythiophene charging. The cathodically colored viologen incorporation into the polythiophene structure was found to be a successful approach for enhancing the over-all EC contrast of the polymeric film. The high coloration efficiency, reasonably low-driving voltage, good thermal stability and excellent switching kinetics made PTh-V an ideal candidate for future optoelectronics and ECDs.Acknowledgements
Financial support from the Academy of Finland is gratefully acknowledged. We would like to thank the Centre of Spectroelectrochemistry, IFW Dresden for providing instrumentation facilities and Frank Ziegs for assistance and fruitful discussion.Notes and references
- P. M. Monk, R. J. Mortimer and D. R. Rosseinsky, Electrochromism: fundamentals and applications, John Wiley & Sons, 2008 Search PubMed.
- P. R. Somani and S. Radhakrishnan, Mater. Chem. Phys., 2003, 77, 117–133 CrossRef CAS.
- R. J. Mortimer, Electrochim. Acta, 1999, 44, 2971–2981 CrossRef CAS.
- K. Hyodo, Electrochim. Acta, 1994, 39, 265–272 CrossRef CAS.
- M. Frechette, M. Belletete, J. Bergeron, G. Durocher and M. Leclerc, Macromol. Chem. Phys., 1997, 198, 1709–1722 CrossRef CAS PubMed.
- M. Leclerc and K. Faid, Adv. Mater., 1997, 9, 1087–1094 CrossRef CAS PubMed.
- M. Leclerc and G. Daoust, Synth. Met., 1991, 41, 529–532 CrossRef.
- L. Robitaille, M. Leclerc and C. Callender, Chem. Mater., 1993, 5, 1755–1761 CrossRef CAS.
- J. Guay, A. F. Diaz, J. Bergeron and M. Leclerc, J. Electroanal. Chem., 1993, 361, 85–91 CrossRef CAS.
- R. Gracia and D. Mecerreyes, Polym. Chem., 2013, 4, 2206–2214 RSC.
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC.
- Y. Long, M. Li, C. Gu, M. Wan, J. Duvail, Z. Liu and Z. Fan, Prog. Polym. Sci., 2011, 36, 1415–1442 CrossRef CAS PubMed.
- P. M. S. Monk, The viologens: physicochemical properties, synthesis, and applications of the salts of 4,4′-bipyridine, Wiley, 1998 Search PubMed.
- B. Gadgil, P. Damlin, T. Ääritalo, J. Kankare and C. Kvarnström, Electrochim. Acta, 2013, 97, 378–385 CrossRef CAS PubMed.
- B. Gadgil, P. Damlin, T. Ääritalo and C. Kvarnström, Electrochim. Acta, 2014, 133, 268–274 CrossRef CAS PubMed.
- L. Beverina, G. A. Pagani and M. Sassi, Chem. Commun., 2014, 50, 5413–5430 RSC.
- L. Dunsch, J. Solid State Electrochem., 2011, 15, 1631–1646 CrossRef CAS PubMed.
- A. Petr, L. Dunsch and A. Neudeck, J. Electroanal. Chem., 1996, 412, 153–158 CrossRef.
- S. Pluczyk, P. Zassowski, R. Rybakiewicz, R. Wielgosz, M. Zagorska, M. Lapkowski and A. Pron, RSC Adv., 2015, 5, 7401–7412 RSC.
- B. Gadgil, E. Dmitrieva, P. Damlin, T. Ääritalo and C. Kvarnström, J. Solid State Electrochem., 2015, 19, 77–83 CrossRef CAS.
- H. C. Ko, S. Park, W. Paik and H. Lee, Synth. Met., 2002, 132, 15–20 CrossRef CAS.
- H. C. Ko, S. Kim, H. Lee and B. Moon, Adv. Funct. Mater., 2005, 15, 905–909 CrossRef CAS PubMed.
- R. D. Webster, R. A. W. Dryfe, J. C. Eklund, C. Lee and R. G. Compton, J. Electroanal. Chem., 1996, 402, 167–174 CrossRef.
- C. Lee, Y. M. Lee, M. S. Moon, S. H. Park, J. W. Park, K. G. Kim and S. Jeon, J. Electroanal. Chem., 1996, 416, 139–144 CrossRef CAS.
- A. Österholm, A. Petr, C. Kvarnström, A. Ivaska and L. Dunsch, J. Phys. Chem. B, 2008, 112, 14149–14157 CrossRef PubMed.
- J. L. Bredas and G. B. Street, Acc. Chem. Res., 1985, 18, 309–315 CrossRef CAS.
- B. Gadgil, P. Damlin, M. Heinonen and C. Kvarnström, Carbon, 2015, 89, 53–62 CrossRef CAS PubMed.
- T. Kuo, C. Hsu, K. Lee and K. Ho, Sol. Energy Mater. Sol. Cells, 2009, 93, 1755–1760 CrossRef CAS PubMed.
- S. Kao, Y. Lin, K. Chin, C. Hu, M. Leung and K. Ho, Sol. Energy Mater. Sol. Cells, 2014, 125, 261–267 CrossRef CAS PubMed.
- J. Lin and X. Ni, RSC Adv., 2015, 5, 14879–14886 RSC.
- T. M. Benedetti, T. Carvalho, D. C. Iwakura, F. Braga, B. R. Vieira, P. Vidinha, J. Gruber and R. M. Torresi, Sol. Energy Mater. Sol. Cells, 2015, 132, 101–106 CrossRef CAS PubMed.
- J. U. Lee, A. Cirpan, T. Emrick, T. P. Russell and W. H. Jo, J. Mater. Chem., 2009, 19, 1483–1489 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra04618a |
| This journal is © The Royal Society of Chemistry 2015 |