
Iterative double cyclization reaction by SRN1 mechanism. A theoretical interpretation of the regiochemical outcome of diazaheterocycles†
Lucas E. Peisino,
Gloria P. Camargo Solorzano,
María E. Budén* and
A. B. Pierini*
INFIQC, Departamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Ciudad Universitaria, 5000 Córdoba, X5000HUA, Argentina. E-mail: eugebuden@yahoo.com.ar; adriana@fcq.unc.edu.ar; Tel: +54-0351-5353867
First published on 13th April 2015
Abstract
In this report, we present a synthetic and mechanistic study of novel iterative double cyclization intramolecular SRN1 reactions from diamides bearing two aryl iodide moieties. This cyclization affords aromatic diazaheterocyclic compounds in good yields. Two synthetic strategies were employed for their preparation: intramolecular SRN1 and Homolytic Aromatic Substitution. The mechanism is non-trivial and we propose that radicals are intermediates. The regiochemistry was studied using computational calculations, employing the DFT method and the B3LYP functional. It was found that the distribution of products depends on the cyclization activation energies, proportion of neutral conformers, and the type of the electron transfer reaction.
1 Introduction
Aromatic azaheterocycles present interesting pharmacological properties related to the planarity of the system and consequently to their DNA-chain intercalating abilities, which make them suitable for anti-neoplastic or mutagenic applications.1–4 Due to their significant biological activity, they are an important class of heterocyclic compounds in medicinal chemistry, being able to bind with high affinities to the aryl hydrocarbon receptor (AhR), which activates the regulatory protein. This effect was studied experimentally and using QSAR methods.5–8In another context, N-containing aromatic heterocycles having more than one nitrogen atom have received an increasing interest owing to the fact that their complexes with transition-metal ions show interesting properties in harvesting light and reemitting it at a wavelength that depends on the metal ion used.9,10
The radical nucleophilic substitution, or SRN1 reaction, is a process through which an aromatic nucleophilic substitution is achieved. Since the scope of this process has been increased considerably over recent decades, it has become an important synthetic strategy.11 The initiation step is by an electron transfer (ET) from suitable donors (i.e., the nucleophile or a base) to the substrate to afford a radical anion. In some systems, the ET step is spontaneous. However, in others, light, electrons from dissolved alkali metals in liquid ammonia, from a cathode or inorganic salts (i.e., Fe2+ or SmI2) are needed to initiate the reaction.12
Several nucleophiles, for example carbanions and heteroatomic anions, can be used for SRN1 reactions to form new C–C or C-heteroatom bonds in good yields. However, an exception to these is the reaction of phenyl amide anions with haloaromatic substrates, where C–N and C–C bond formations were achieved instead.13 2-Naphthylamide anions can react by the photo SRN1 process with PhI, 4-MeOC6H4I and 1-iodonaphthalene in liquid ammonia. Here, 1-aryl 2-naphthylamines were formed regioselectively in 45–63% yields, with only 3–6% of N-arylation.14 Moreover, double arylation has been previously achieved using p-dihalobenzene as a substrate with the anions of 2-naphthylamine and 9-phenanthrylamine under irradiation in liquid ammonia.15
An SRN1 synthetic strategy to obtain heterocyclic compounds was previously developed based on the intramolecular cyclization of substrates bearing both the leaving group and the nucleophilic center.16 This methodology has been recently applied to the synthesis of 1-phenyl-1-oxazolinoindan derivatives and their related compounds;17 tetracyclic isoquinoline derivatives;18 a series of substituted 9H-carbazoles and carbolines;19–21 aporphine and homoaporphine alkaloids;22 pyrroles, indoles, and pyrazoles;23 indazoles;24 pyridio[1,2-a]benzimidazoles;25 2-pyrrolyl and 2-indolyl benzoxazoles,26,27 among others. Moreover, an intermolecular SRN1 reaction of substituted iodobenzylamines with several tetralones afforded a series of benzo[c]phenanthridines with modest overall yields after several steps.28
Recently, Rossi et al. proposed a new approach for the syntheses of phenanthridines and benzophenanthridines (2) by intramolecular ortho-arylation of (2-halobenzyl)-N-arylamines (1) (eqn (1)).29,30 In these reactions, the cyclization of compounds such as 1 gives very good yields of the phenanthridine derivates 2 (44–85%). On other hand, by direct photolysis of the C–I bond of 1, the synthesis of 2 was achieved with 30–95% yields via Homolytic Aromatic Substitution (HAS).31,32
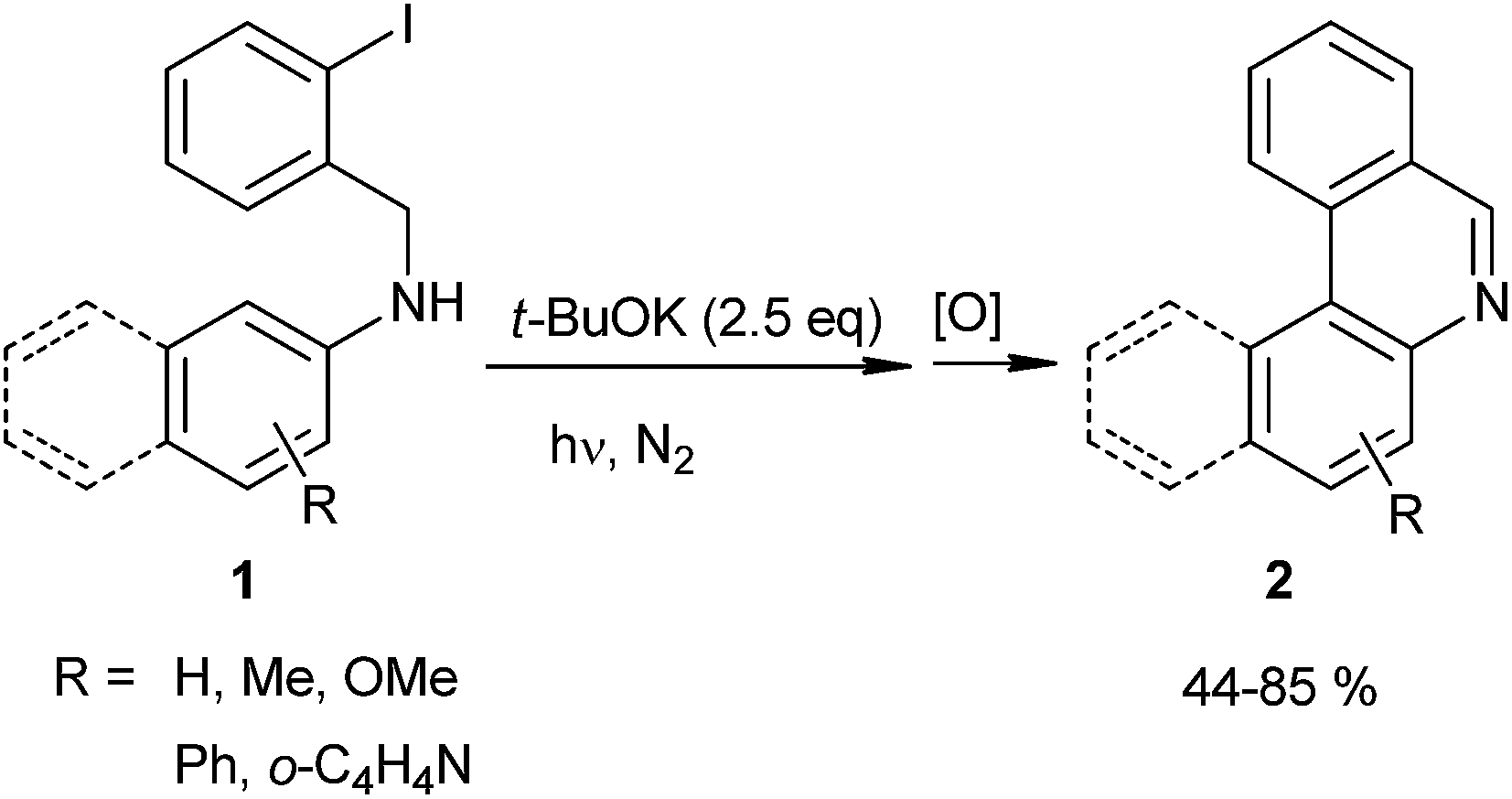 | (1) |
It is worth noting in the literature an example reported of double ring closure reactions by SRN1 where two 5-membered rings are formed.19 In this case, two intramolecular consecutive SRN1 of substrate 3 were reported to give dicarbazol 4 in 67% yield (eqn (2)). However, there is no other reported example where a double closure afforded two 6-membered rings. With this in mind, in the present article we report a double cyclization reaction to give 6-membered diazaheterocyclic compounds. In order to explain their regiochemical outcome, a computational study of the non-trivial mechanism is presented.
 | (2) |
2 Results and discussion
The substrates were prepared through a three-step synthetic strategy (Scheme 1). The first reaction was the formation of acetamide derivative 6 from 1,4-diaminobenzene (5) with an excess of acetic anhydride. The dianion of amide 6 was formed with NaH (2 equiv.), after which o-iodobenzyl chloride (2 equiv.) was added and yielded the diacetylated dibenzyl amide 7. Finally, acid hydrolysis of the acetamide group was carried out and afforded 8 in a 53% global yield. | ||
| Scheme 1 Synthesis of diamine 8. | ||
Following the same procedure, substrates 9 and 11 were obtained in 26% and 65% isolated yields, from 4,4′-diaminobiphenyl and 1,3-diaminopyridine, respectively (Scheme 2), using the new synthetic intermediates 10 and 12.
 | ||
| Scheme 2 Retrosynthetic preparation of diamines 9 and 11. | ||
2.1 Intramolecular SRN1 reactions
The results of the photostimulated reaction (180 min) of the diamines 8, 9 and 11 in the presence of excess t-BuOK (5 equiv.) under a nitrogen atmosphere are presented in Table 1. Under these reaction conditions, and after oxidation with MnO2/CHCl3, the diamine 8 afforded good yields of diphenanthridines 13 (46%) and 14 (25%) using liquid ammonia as the solvent (Table 1, entries 1–3 and eqn (3)).
 | (3) |
| Entry | Substr. | Solvent | Product | Yields (%) | I− (%)b |
|---|---|---|---|---|---|
| a Photostimulated reactions were performed with [substrate] = 1.33 × 10−3 M and five times with [t-BuOK]. The substrate was previously dissolved in DMSO. Reaction times were 180 min, unless indicated otherwise. Irradiation was conducted in a reactor equipped with two high pressure lamps of model Phillips HPI-T plus 400 W (air- and water-refrigerated) with a maximum emission at 530 nm. Oxidation reactions were carried out by stirring the crude reaction with MnO2 in CHCl3. Yields of the products was determined by 1H-NMR.b The halides were determined potentiometrically.c The substrate was not dissolved in DMSO.d Isolated yield.e Dark conditions. Only the substrate was detected (95% yield).f m-Dinitrobenzene was added (30 mol%).g TEMPO was added (30 mol%).h [Substrate] = 100 × 10−3 M and [t-BuOK] = 70 × 10−2 M.i [Substrate] = 20 × 10−3 M and [t-BuOK] = 140 × 10−3 M.j Reaction time 240 min. Substrate was detected (48% yield). | |||||
| 1 | 8 | NH3(l) | 13 (42) | 14 (20) | 88 |
| 2c | 8 | NH3(l) | 13 (46) | 14 (25) | 91 |
| 3 | 8 | NH3(l) | 13 (46)d | 14 (17)d | 88 |
| 4e | 8 | NH3(l) | 13 (—) | 14 (—) | <5 |
| 5f | 8 | NH3(l) | 13 (3) | 14 (<1) | 32 |
| 6g | 8 | NH3(l) | 13 (18) | 14 (5) | 79 |
| 7h | 8 | DMF | 13 (21) | 14 (8) | 73 |
| 8i | 8 | Diglyme | 13 (11) | 14 (5) | 90 |
| 9j | 9 | NH3(l) | 18 (17) | 42 | |
| 10 | 11 | NH3(l) | 19 (13) 20 (13) 21 (22) | 94 | |
In dark conditions, there was no reaction of 8 with the excess of t-BuOK in liquid ammonia (Table 1, entry 4). The reaction was partially inhibited by m-dinitrobenzene (m-DNB), a well-known inhibitor of the SRN1 processes (Table 1, entry 5), as well as being inhibited by radical traps such as TEMPO (Table 1, entry 6). This indicates, for this system, that the double cyclization is slow with respect to the single ring closures previously reported, in which TEMPO caused no inhibition.29,30
The reaction was tested in the organic solvents DMF, diglyme and DMSO. However, the yields of diphenanthridines 13 and 14 were lower than in NH3(l), due to the greater hydrogen-donor capacity of the media. For instance in DMF, 13 and 14 were formed in 21% and 8%, respectively (entry 7). Using diglyme as the solvent, yields were 11% of 13 and 5% of 14 (entry 8), but only 5% of 13 being obtained in DMSO.‡
In the HPLC/MS chromatogram profile of the selected reaction (Table 1, entry 7; see ESI†), the double-cyclization products 13 and 14 were observed, together with the monocyclization–reduction product 15, monocyclization with iodo retention 16 and the monocyclization–fragmentation (Cbenzylic-N) product 17 (Fig. 1). The absence of a double-reduction product and the presence of 15 and 16 indicate that the first cyclization was favored with respect to the second one. The second cyclization reaction competed with the hydrogen–abstraction from the solvent to afford 15, and with the Cbenzylic-N fragmentation to give 17.
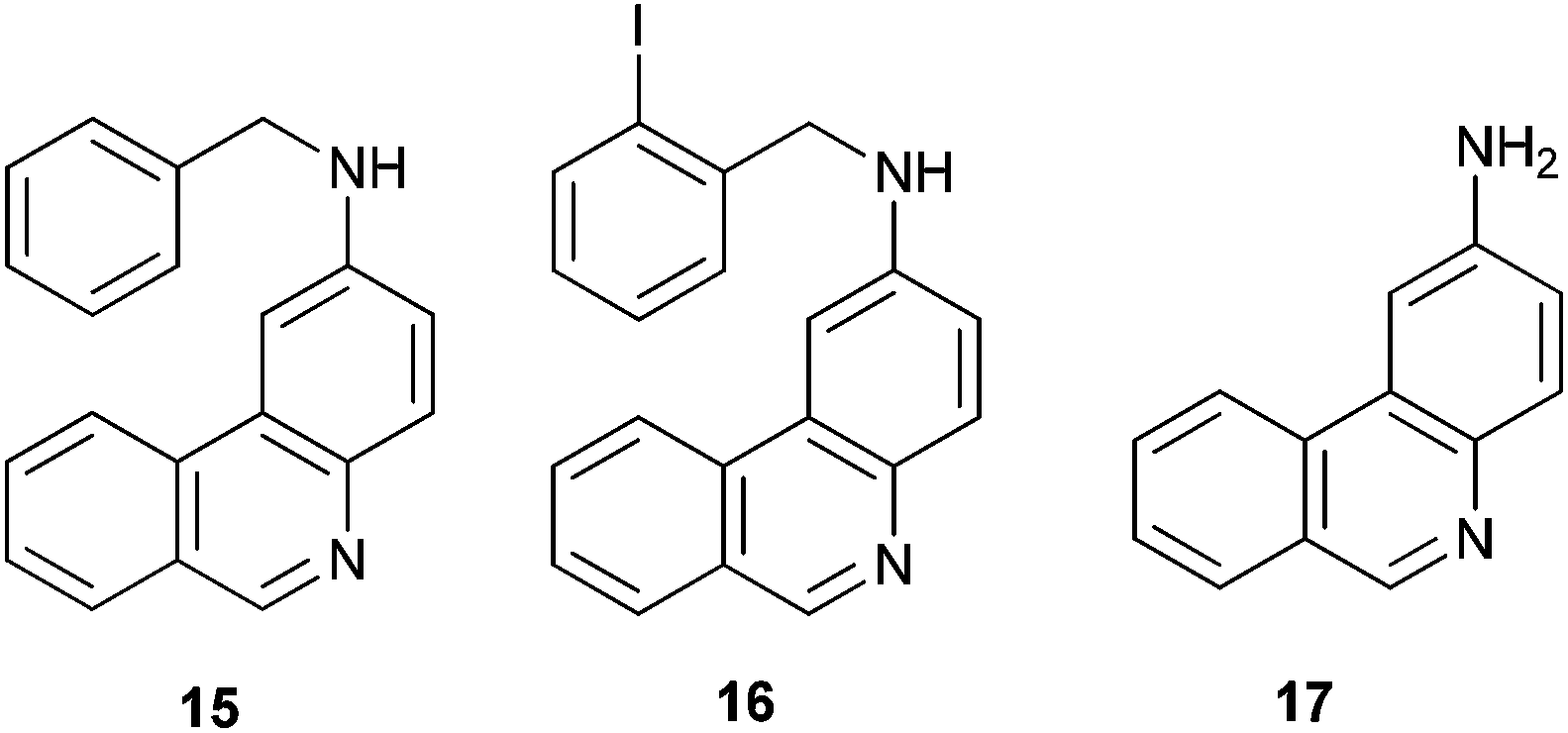 | ||
| Fig. 1 Other products obtained in cyclization reaction of diamine 8. | ||
In this work, the product yield of 13 was 46% (24% overall yield) which is comparable, and in some cases better, than those obtained by other synthetic strategies. Starting from analog chloride derivative type 8, a 4% yield of 13 was obtained through a benzyne type mechanism using NaNH2/THF33 and 40% yield using KNH2/NH3(liq).34 However, there is no specification for the preparation of the chloride derivative type 8, which is not a single molecule. Furthermore, a comparable yield of 13 (49% yield) was also reported using photochemical methodology. However, these reactions were irradiated for 24–36 hours.35
Concerning the synthesis of 14, there is only one report in which, after four consecutive reactions, the product was obtained in 14% yield.36 This value is comparable to that obtained by the route where 14 was afforded with 9% overall yield.
Following the same methodology, the photostimulated reaction of 9 was carried out in NH3(l) as the solvent, and the diphenanthridine 18 was obtained in 17% yield (Table 1, entry 9 and eqn (4)). In this case, a 48% yield of unreacted substrate 9 was achieved, due to its very low solubility in the reaction media. When the reaction was carried out in organic solvents such as diglyme and DMF, no diazaheterocycle 18 was formed, with only a reduced product being observed (results not tabulated). Despite its regular yield, diazaheterocycle 18 has not been described in the literature.
 | (4) |
In the same way, the photostimulated reaction of diamine 11 was carried out in NH3(l), and the double cyclization products 19 (C–C coupling) and 20 (C–N coupling) were both afforded in 13%, together with the monocyclization–reduction product 21 in 22% yield (Table 1, entry 10 and eqn (5)). However, when the reaction was carried out in DMF as the solvent, the yields of the cyclic products 19 and 20 did not improve (results not tabulated).
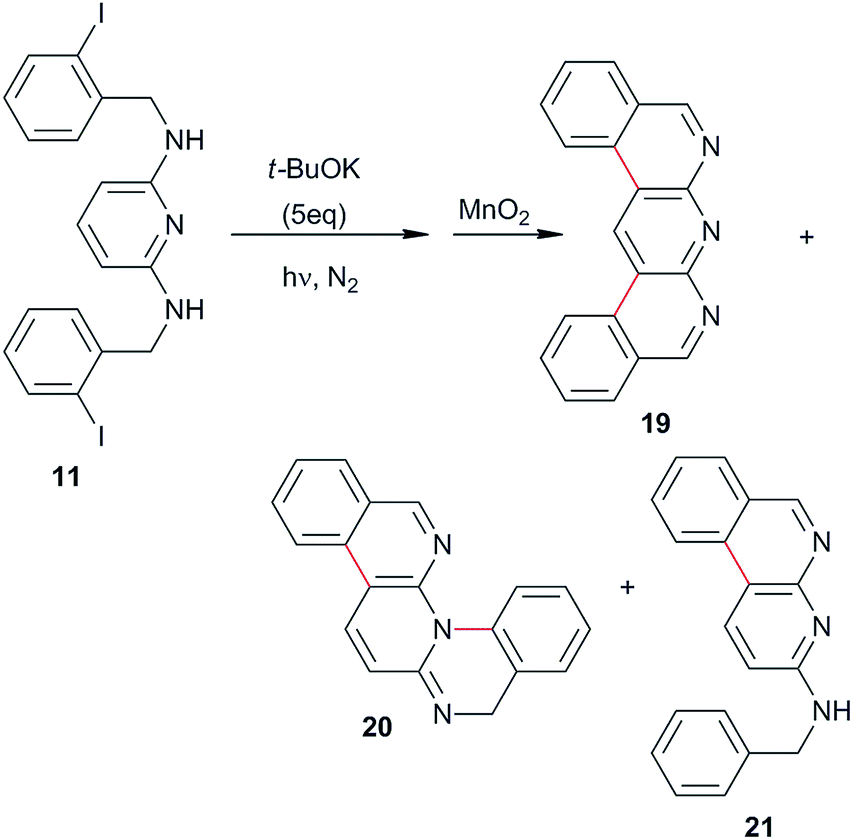 | (5) |
Although, diazaheterocycle 19 was obtained in 68% yield in two consecutive reactions (diiodination of 2,6-diaminopyridine followed by a double Suzuki coupling with 2-formylphenyl boronic acid and spontaneous cyclization and aromatization),37 our strategy did not include the use of a transition metal. This is important because impurities can be avoided and the products used directly in pharmaceutical and other industries. In addition, for compounds 20 and 21 there are no precedent in the literature of their preparation.
Taking into account that in all reactions double cyclization products were formed, and considering the results presented in Table 1 and those previously reported,29,30 we suggest that SRN1 is the operating mechanism. Furthermore, the results in dark conditions, the inhibition exerted by TEMPO and another inhibitor (m-DNB), and the presence of monocyclization–reduction product 15, are consistent with aryl radicals and radical anions as intermediates.26,27
2.2 Intramolecular HAS reactions
In order to explore other possible reaction patterns of the substrates described above, we used the direct photolysis reaction of compounds 8, 9 and 11 to obtain the diazaheterocycles. These reactions were carried out in a quartz tube using anhydrous acetonitrile as the solvent. Photostimulation was achieved by irradiating with a wavelength of 254 nm. In this cyclization reaction, the presence of a base is not required because initiation takes place through the homolytic C–I breaking bond. The aryl radicals thus formed can be added to the π system of the central aromatic ring to yield a cyclohexadienyl radical. Possibly, this radical transfers the tertiary hydrogen atom to other radical intermediates present in the reaction media (i.e. iodine atom) to give the neutral cyclic product.31After a screening of conditions,§ the best results were found to be obtained when degassing the reaction media by nitrogen bubbling and sonication. Results of photolysis reactions in these conditions for the three substrates studied are presented in Table 2. In the photostimulated reaction (120 minutes) of 8, the cyclic product 13 was obtained in 36% yield, and traces of isomer 14 were also obtained (Table 2, entry 2). Although the SRN1 reaction gave a higher yield of both products (Table 1, entry 2), the direct photolysis was more selective towards product 13.
| Entry | Substrate | Time (min) | Yield (%) |
|---|---|---|---|
| a Photostimulated reactions were performed with [substrate] = 1 mg mL−1 with acetonitrile as solvent (7 mL) in a quartz tube, under a nitrogen atmosphere and bubbling during all the reaction time. The reaction mixture was previously degassed by nitrogen bubbling and sonication for 20 minutes. Irradiation was conducted in a photochemical reactor equipped with nine Hg high pressure lamps (254 nm). Oxidation reactions were carried out by stirring the crude reaction with MnO2 in CHCl3. Product yields were determined by 1H-NMR. | |||
| 1 | 8 | 60 | 13 (28) 14 (<5) |
| 2 | 8 | 120 | 13 (36) 14 (5) |
| 3 | 9 | 60 | 18 (6) |
| 4 | 9 | 120 | 18 (6) |
| 5 | 9 | 180 | 18 (6) |
| 6 | 11 | 60 | 19 (—) 20 (—) 21 (32) |
In contrast with the SRN1 conditions (Table 1, entry 9), substrate 9 was completely soluble in the direct photolysis conditions. However, the yield of the double-cyclization product 18 was less than 10% (Table 2, entries 3–5). A similar behavior was observed in the reaction of 11, where the double-cyclization products (19 and 20) were not obtained and only the monocyclization–reduction product 21 was detected (Table 2, entry 6).
2.3 Mechanism and theoretical calculations
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for 8 s-cis:8 s-trans.
:1 for 8 s-cis:8 s-trans.
 | (6) |
In the superbasic reaction medium, the dianions 8 s-cis2− and 8 s-trans2− can be formed. The s-cis/s-trans isomerization barrier for the anions is high, because it involves rotation around C3(phenyl) and the N2 bond, which has a partial double-bond character. Thus, the distribution of the anions 8 s-cis2−:8 s-trans2− is directly related to its neutrals (8 s-cis:8 s-trans).
The initiation step involves a photoinduced ET to 82− followed by fragmentation of a C–I bond to give the distonic radical dianion¶ [8−]˙− (s-cis and s-trans) and I− anion (Scheme 3). The intermediate radical dianion [8−]˙− adds quickly, via intramolecular C–C cyclization, to afford the monocyclic conjugated radical dianion [22−]˙− (s-cis and s-trans), which is separated from the second o-iodoaryl moiety by a Csp3 atom. The radical dianion [22−]˙− may follow either an intramolecular (Scheme 4) or an intermolecular ET reaction pathway (Scheme 5).
 | ||
| Scheme 3 Possible initiation step and first ring closure reaction of 82−. | ||
 | ||
| Scheme 4 Intramolecular ET from [22−]˙−. | ||
 | ||
| Scheme 5 Intermolecular ET from [22−]˙−. | ||
Following the intramolecular pathway, 23˙− (s-cis and s-trans) can be formed (Scheme 4). However, cyclization to give radical anion 24˙− is favored from 23˙− s-cis because in 23˙− the negative charge is localized in the C vecinal to Csp3 of the central ring (Fig. 2). After ring closure radical anion 24˙− is yielded. This transfers the extra electron to 82− to afford 24 and [8−]˙−, with the latter propagating the reaction cycle. Ultimate tautomerization of 24 in the basic media, and subsequent oxidation will afford 13. Thus, the 23˙− s-trans isomer cannot cyclize, and reduction of this radical anion by hydrogen-abstraction from the solvent occurs to finally yield the monocyclization–reduction product 15 (Scheme 4). Under this intramolecular ET pathway, only the cis product 13 and reduced product 15 will be observed.
 | ||
| Fig. 2 (A) Resonance structures for radical anion 23˙−. (B) Electrostatic potential of radical anions 23˙− and [26−]˙−. | ||
On the other hand, if intermolecular ET from [22−]˙− to 82− is followed, 22− (s-cis and s-trans) and [8−]˙− can be afforded (Scheme 5). The anion 22− in the basic medium rearomatizes the central ring to give 252− (s-cis and s-trans), which can initiate a second SRN1 cycle through the formation of the distonic radical anion [26−]˙− (s-cis and s-trans). Given the electronic distribution of [26−]˙−, both ortho positions to the amide group are favored to couple with aryl radicals (Fig. 2B). Following this reactive pathway, [26 s-cis−]˙− and [26 s-trans−]˙− can cyclize to give products 13 and 14. Experimentally, we observed that both products are formed together with products 15 and 16 (Fig. 1). Moreover, product 16 is formed through intermediate 252−, via protonation–oxidation reactions. Therefore, intermolecular ET is operating in the cyclization of 82−.
Considering that the electronic structure of radical dianion [26−]˙− (s-cis and s-trans) corresponds to a conjugated species and that the evaluated energy for its cyclization is high (see ESI†), we propose that cyclization to occurs via the distonic radical anion 26˙− (s-cis and s-trans), which can be present under the reaction conditions (eqn (7)).
 | (7) |
The PES for cyclization of radical anions 26˙− (s-cis and s-trans) presented in Fig. 3 shows that both conformers cyclize with similar energies to give the s-cis and s-trans cyclic products. Considering that the kinetics of ring closure are similar and that the interconversion between the s-cis and s-trans conformers is very slow because it implies a rotation around C–N bond with a double-bond character,|| then the product distribution for this path is governed by this conformational distribution and should be close to 1.2:1. Taking into account that the experimental ratio of s-cis:s-trans products is 1.8:1, we propose that not only the intermolecular ET pathway, but also the intramolecular ET pathway is taking place in this system.
 | ||
| Fig. 3 PES to cyclization of 26˙− radical anion. | ||
:1.8, respectively (T = 240 K).
 | (8) |
As mentioned above, the presence of product 21 (see enq (5)) indicate that the first cyclization (C–C coupling) is favored with respect to the second one (C–C or C–N coupling).
In principle, once radical dianion [28−]˙− is formed, two reactive pathways may be followed: intramolecular ET (Scheme 6) and intermolecular ET (Scheme 7).
 | ||
| Scheme 6 Intramolecular ET from [28−]˙−. | ||
 | ||
| Scheme 7 Intermolecular ET from [28−]˙−. | ||
Similar to 8, if the reaction takes place by intramolecular ET (Scheme 6), only product 20 (C–N coupling) will be obtained from the 29 s-trans˙−. This behavior can be explained due to the negative charge being localized on the N atom of the pyridine central ring for the radical anion 29˙− (Fig. 4). Thus, the 29˙− s-cis isomer cannot cyclize, and reduction of this radical anion (by hydrogen-abstraction from the solvent) will occur to finally yield the monocyclization–reduction product 21. Under this intramolecular pathway, product 19 will not be formed.
 | ||
| Fig. 4 (A) Resonance structures for radical anion 29˙−. (B) Electrostatic potential of radical anions 29˙− and [31−]˙−. | ||
If intermolecular ET is in play (Scheme 7), the formation of both products 19 and 20 is possible. As the relationship of 112− s-cis:s-trans conformers is 1:1.8, the ratio of the radical anions 32 s-cis˙− to 32 s-trans˙− will remain constant as well as the ratio of products.**
Taking this into account, the second cyclization path was theoretically investigated. The PES for this system shows that the activation energy for C–N coupling is 10.4 kcal mol−1, while for the C–C coupling is only 2.0 kcal mol−1 (Fig. 5). On the other hand, the activation energy for hydrogen-abstraction of [32−]˙− to yield 21 is 6.5 kcal mol−1. This indicates that hydrogen-abstraction competes with C–N coupling, and thus only the monocyclation–reduction product 21 is obtained from [32 s-trans−]˙− and only product 19 is formed from [32 s-cis−]˙−. Accordingly, product 20 is formed by the intramolecular ET pathway (Scheme 6). Similarly to 8, in this system both ET pathways are present.
 | ||
| Fig. 5 PES to cyclization of radical anion 32˙−. | ||
3 Conclusions
In this article, we have presented the synthesis of novel diazaheterocycles by double intramolecular SRN1 reactions. This is the first study that explores in detail reactions where two ring closures occur consecutively. Considering that during the reaction two new junctions C–C or C–N are formed to result in the formation of two new heterocycles within the same molecule, it is ensured that the diazaheterocycles have good yields (13, 46%; 14, 25%; 18, 17%; 19, 13% and 20, 13%). Direct photolysis reactions of the amines 8, 9 and 11 were carried out and dual-closure rings were observed. However, a lower percentage of products than that obtained by SRN1 were found. This may have been due to the instability of intermediates and products formed under the irradiation conditions used in the photolysis reactions.Radicals and radical anions are intermediates of these reactions. The mechanism in the presence of a base was studied using DFT calculations, with the product distribution depending not only on the ratio of conformers of the neutral species, but also on the type of the ET reaction and the relative energies of the coupling. After the first cyclization, the reduction of the aryl radical was always in competition with the second cyclization reaction. Finally, we suggest that for these systems both intramolecular and intermolecular ET were present.
4 Experimental
4.1 Computational procedure
The conformational search was carried out using a Vconf program.†† Calculations were performed using the Gaussian09 program, the B3LYP38,39 DFT functional and the 6-31++G* basis set. The B3LYP functional and the 6-31+G*,30,40 6-311+G*,21,41 and 6-311+G**40 basis sets have been previously tested for similar systems. All determinations were carried out with full geometry optimization, including for all cases the effect of the solvent through Tomasi's polarized continuum model (PCM)42–44 as implemented in Gaussian09. The effect of NH3(l) was evaluated using methanol as the model polar solvent.30 The TS and intermediates were localized by a scan of the distinguished reaction coordinate. Then, after refinement, a characterization of stationary points was made by Hessian matrix calculations, with all positive eigenvalues for a minimum and only one negative eigenvalue for the TSs. The energy reported for all species includes zero-point corrections.4.2 General methods
The products were quantified by 1H-NMR. All NMR spectra were obtained on a 400 MHz Spectrometer (1H-NMR (400 MHz), 13C-NMR (100 MHz), COSY, HSQC, HMBC and NOE) using CDCl3 as the solvent unless otherwise indicated. The coupling constants (J) are given in hertz. The HPLC/MS analyses were carried out on a HPLC equipment with a reverse C-18 stationary phase (15 cm × 4.6 × 5 micron) and MeCN–water mixtures as the mobile phase; coupled to high-resolution mass spectra on a TOF analyzer, using an ESI† ion source, with nitrogen as the nebulizing and drying gas. High-resolution mass spectra were recorded on a TOF analyzer, using an ESI† source in a positive mode, with nitrogen as the nebulizing and drying gas, and sodium formate (10 mM) as the internal calibrant.4.3 Materials
Acetic anhydride, sodium hydride (60% on mineral oil), 2-iodobenzyl chloride and potassium t-butoxide were obtained from commercial sources. DMSO was stored over 4 Å molecular sieves. DMF was distilled from Na metal and stored under nitrogen over 4 Å molecular sieves. Diglyme was distilled from Na metal and stored with Na wires. To prepare the substrates, commercially available 1,4-diaminobenzene, 4,4′-diaminobiphenyl and 2,6-diaminopyridine were used. Silica gel (0.063–0.200 mm) was used in column chromatography and on 2 mm plates (silica gel 60 PF254) in radial thin-layer chromatography purification. All solvents were of analytical grade and used as received from the supplier.The compound 8 was isolated by crystallization as hydrochloride from acid ethanol and basic medium extraction. 1H-NMR (400 MHz, CDCl3), δH: 3.82 (br s, 2H); 4.24 (s, 4H); 6.53 (s, 4H); 6.95 (td, 2H, J = 7.5 Hz, 1.6 Hz); 7.29 (td, 2H, J = 7.5 Hz, 1.1 Hz); 7.39 (dd, 2H, J = 7.7 Hz, 1.6 Hz); 7.83 (dd, 2H, J = 7.9 Hz, 1.1 Hz). 13C-NMR (100 MHz, CDCl3) δC: 54.3; 98.6; 114.8; 128.4; 128.8; 129.0; 139.4; 140.3; 141.5. ESI-HRMS m/z [M + H]+ calcd for C20H19I2N2 540.9632, found 540.9638.
:dichloromethane, 30:70. 1H-NMR (400 MHz, CDCl3), δH: 4.21 (br s, 2H); 4.35 (s, 4H); 6.64 (br d, 4H, J = 6.5 Hz); 6.98 (td, 2H, J = 7.6 Hz, 1.6 Hz); 7.31 (td, 2H, J = 7.6 Hz, 1.1 Hz); 7.35 (br d, 4H, J = 8.0 Hz); 7.1 (dd, 2H, J = 7.7 Hz, 1.6 Hz); 7.86 (dd, 2H, J = 7.9 Hz, 1.1 Hz). 13C-NMR (100 MHz, CDCl3) δC: 53.4; 98.5; 113.3; 127.2; 128.4; 128.8; 129.0; 131.1; 139.5; 141.0; 146.2. ESI-HRMS m/z [M + H]+ calcd for C26H23I2N2 616.9945, found 616.9950.:ethyl acetate, 80:20. 1H-NMR (400 MHz, CDCl3), δH: 7.55 (ddd, 2H, J = 8.5 Hz, 7.0 Hz, 1.4 Hz); 7.67 (ddd, 2H, J = 8.0 Hz, 7.0 Hz, 1.0 Hz); 8.13 (dd, 2H, J = 8.0 Hz, 0.8 Hz); 8.29 (s, 2H); 8.51 (d, 2H, J = 8.5 Hz); 9.43 (s, 2H). 13C-NMR (100 MHz, CDCl3) δC: 120.1; 126.6; 127.0; 127.6; 128.1; 129.0; 130.8; 132.9; 145.1; 153.4. ESI-HRMS m/z [M + H]+ calcd for C20H13N2 281.1073, found 281.1085.:ethyl acetate, 80:20. 1H-NMR (400 MHz, CDCl3), δH: 7.78 (t, 2H, J = 7.5 Hz); 7.94 (td, 2H, J = 7.0 Hz, 1.1 Hz); 8.07 (d, 2H, J = 7.9 Hz); 8.79 (d, 2H, J = 8.2 Hz); 9.32 (s, 2H); 9.33 (s, 2H). 13C-NMR (100 MHz, CDCl3) δC: 122.5; 123.4; 124.8; 126.2; 128.2; 129.0; 131.5; 132.4; 142.6; 154.9. ESI-HRMS m/z [M + H]+ calcd for C20H13N2 281.1073, found 281.1065.:methanol, 98:2. 1H-NMR (400 MHz, CDCl3), δH: 7.76 (t, 2H, J = 7.4 Hz); 7.92 (td, 2H, J = 7.7 Hz, 1.1 Hz); 8.10 (d, 2H, J = 7.9 Hz); 8.16 (dd, 2H, J = 8.5 Hz, 1.9 Hz); 8.33 (d, 2H, J = 8.5 Hz); 8.76 (d, 2H, J = 8.3 Hz); 8.92 (d, 2H, J = 1.6 Hz); 9.32 (s, 2H). 13C-NMR (100 MHz, CDCl3) δC: 121.1; 122.0; 124.5; 126.7; 127.8; 128.4; 129.0; 130.8; 131.2; 132.6; 139.6; 144.1; 153.8. ESI-HRMS m/z [M + H]+ calcd for C26H17N2 357.1386, found 357.1399.:methanol, 99:1 to 91:9. 1HNMR (400 MHz, CDCl3), δH: 7.87 (t, 2H, J = 7.5 Hz); 8.05 (t, 2H, J = 7.6 Hz); 8.20 (d, 2H, J = 7.8 Hz); 8.90 (d, 2H, J = 8.3 Hz); 9.67 (s, 2H); 10.17 (s, 1H). 13C-NMR (100 MHz, CDCl3) δC: 118.5; 122.4; 126.3; 127.4; 128.9; 129.6; 132.1; 132.4; 153.7; 159.7. ESI-HRMS m/z [M + Na]+ calcd for C19H11N3Na 304.0848, found 304.0851.:methanol, 98:2. 1H-NMR (400 MHz, acetone-d6), δH: 5.30 (s, 2H); 7.00 (br t, 1H, J = 7.5 Hz); 7.28 (d, 1H, J = 7.2 Hz); 7.338 (br d, 1H, J = 8.6 Hz); 7.340 (br t, 1H, J = 7.9 Hz); 7.43 (br d, 1H, J = 7.6 Hz); 7.71 (br t, 1H, J = 7.1 Hz, 7.5 Hz); 7.92 (cplx. t, 1H, J = 7.7 Hz); 8.22 (br d, 1H, J = 8.1 Hz); 8.67 (br d, 1H, J = 8.3 Hz); 9.04 (d, 1H, J = 8.9 Hz); 9.47 (s, 1H). 13C-NMR (from HSQC and HMBC, acetone-d6) δC: 59.6; 109.0; 109.4; 112.0; 120.9; 121.0; 121.3; 125.3; 126.0; 128.0; 128.7; 130.9; 131.3; 133.5; 151.3; 153.9; 154.0; 155.2; 156.3. ESI-HRMS m/z [M + H]+ calcd for C19H14N3 284.1182, found 284.1191.:methanol, 98:2. 1HNMR (400 MHz, CDCl3), δH: 4.81 (d, 2H, J = 5.4 Hz); 5.81 (br s, 1H); 6.81 (d, 1H, J = 8.9 Hz); 7.30 (br d, 1H, J = 7.3 Hz); 7.35 (br t, 2H, J = 7.3 Hz); 7.44 (br d, 2H, J = 7.7 Hz); 7.59 (td, 1H, J = 7.1 Hz, 0.8 Hz); 7.80 (ddd, 1H, J = 8.3 Hz, 7.0 Hz, 1.3 Hz); 8.02 (br d, 1H, J = 8.0 Hz); 8.34 (d, 1H, J = 8.4 Hz); 8.57 (d, 1H, J = 8.9 Hz); 9.36 (s, 1H). 13C-NMR (100 MHz, CDCl3) δC: 46.1; 110.4; 111.1; 120.7; 125.1; 125.9; 127.5; 127.8; 128.8; 129.0; 131.3; 133.1; 133.4; 138.8; 153.5; 156.4; 158.3. ESI-HRMS m/z [M + H]+ calcd for C19H16N3 286.1339, found 286.1352.Acknowledgements
This work was supported in part by the Consejo Nacional de Investigaciones Cientificas y T'ecnicas (CONICET), the Agencia Nacional de Promoción Científica y Técnica (ANPCyT) and the Secretaria de Ciencia y T'ecnologia (Universidad Nacional de Córdoba), Argentina. L.E.P and G.P.C.S. gratefully acknowledge the receipt of a fellowship from CONICET.Notes and references
- B. P. Hudson and J. K. Barton, J. Am. Chem. Soc., 1998, 120, 6877–6888 CrossRef CAS.
- S. W. Fewell and J. L. J. Woolford, Mol. Cell. Biol., 1999, 19, 826–834 CAS.
- H.-L. Chan, H.-Q. Liu, B.-C. Tzeng, Y.-S. You, S.-M. Peng, M. Yang and C.-M. Che, Inorg. Chem., 2002, 41, 3161–3171 CrossRef CAS PubMed.
- C. Bailly, Curr. Med. Chem., 2000, 7, 39–58 CrossRef CAS.
- E. Lo Piparo, K. Koehler, A. Chana and E. Benfenati, J. Med. Chem., 2006, 49, 5702–5709 CrossRef CAS PubMed.
- C. L. Waller and J. D. McKinney, Chem. Res. Toxicol., 1995, 8, 847–858 CrossRef CAS.
- M. Gillner, J. Bergman, C. Cambillau, M. Alexandersson, B. Fernström and J. A. Gustafsson, Mol. Pharmacol., 1993, 44, 336–345 CAS.
- U. Rannug, M. Sjögren, A. Rannug, M. Gillner, R. Toftgard, J. A. Gustafsson, H. Rosenkranz and G. Klopman, Carcinogenesis, 1991, 12, 2007–2015 CrossRef CAS.
- K. E. S. Phillips, T. J. Katz, S. Jockusch, A. J. Lovinger and N. J. Turro, J. Am. Chem. Soc., 2001, 123, 11899–11907 CrossRef CAS PubMed.
- D. Waghray, A. Cloet, K. Van Hecke, S. F. L. Mertens, S. De Feyter, L. Van Meervelt, M. Van der Auweraer and W. Dehaen, Chem.–Eur. J., 2013, 19, 12077–12085 CrossRef CAS PubMed.
- J. I. Bardagí, V. A. Vaillard and R. A. Rossi, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, John Wiley & Sons, Ltd, 2012, ch. The SRN1 Reaction, pp. 1505–1519 Search PubMed.
- R. A. Rossi, A. B. Pierini and A. B. Peñéñory, Chem. Rev., 2003, 103, 71–167 CrossRef CAS PubMed.
- J. F. Bunnett and J. K. Kim, J. Am. Chem. Soc., 1970, 92, 7464–7466 CrossRef CAS.
- A. B. Pierini, M. T. Baumgartner and R. A. Rossi, Tetrahedron Lett., 1987, 28, 4653–4656 CrossRef CAS.
- L. B. Jimenez, N. V. Torres, J. L. Borioni and A. B. Pierini, Tetrahedron, 2014, 70, 3614–3620 CrossRef CAS PubMed.
- R. A. Rossi and M. T. Baumgartner, Synthesis of Heterocycles by the SRN1 Mechanism in Targets in Heterocyclic System: Chemistry and Properties, Soc. Chimica. Italiana, Rome, Italy, 1999, vol. 3, pp. 215–243 Search PubMed.
- L. J. Marshall, M. D. Roydhouse, A. M. Z. Slawin and J. C. Walton, J. Org. Chem., 2007, 72, 898–911 CrossRef CAS PubMed.
- M. D. Roydhouse and J. C. Walton, Eur. J. Org. Chem., 2007, 1059–1063 CrossRef CAS PubMed.
- M. E. Budén, V. A. Vaillard, S. E. Martín and R. A. Rossi, J. Org. Chem., 2009, 74, 4490–4498 CrossRef PubMed.
- J. K. Laha, S. M. Barolo, R. A. Rossi and G. D. Cuny, J. Org. Chem., 2011, 76, 6421–6425 CrossRef CAS PubMed.
- W. D. Guerra, R. A. Rossi, A. B. Pierini and S. M. Barolo, J. Org. Chem., 2015, 80, 928–941 CrossRef CAS PubMed.
- S. M. Barolo, X. Teng, G. D. Cuny and R. A. Rossi, J. Org. Chem., 2006, 71, 8493–8499 CrossRef CAS PubMed.
- V. A. Vaillard, M. E. Budén, S. E. Martín and R. A. Rossi, Tetrahedron Lett., 2009, 50, 3829–3832 CrossRef CAS PubMed.
- I. Thomé, C. Besson, T. Kleine and C. Bolm, Angew. Chem., Int. Ed., 2013, 52, 7509–7515 CrossRef PubMed.
- S. M. Barolo, Y. Wang, R. A. Rossi and G. D. Cuny, Tetrahedron, 2013, 69, 5487–5494 CrossRef CAS PubMed.
- V. A. Vaillard, J. F. Guastavino, M. E. Budén, J. I. Bardagí, S. M. Barolo and R. A. Rossi, J. Org. Chem., 2012, 77, 1507–1519 CrossRef CAS PubMed.
- V. A. Vaillard, R. A. Rossi and J. E. Argüello, Org. Biomol. Chem., 2012, 10, 9255–9261 CAS.
- R. Beugelmans, J. Chastanet, H. Ginsburg, L. Quintero-Cortes and G. Roussi, J. Org. Chem., 1985, 50, 4933–4938 CrossRef CAS.
- M. E. Budén and R. A. Rossi, Tetrahedron Lett., 2007, 48, 8739–8742 CrossRef PubMed.
- M. E. Budén, V. B. Dorn, M. Gamba, A. B. Pierini and R. A. Rossi, J. Org. Chem., 2010, 75, 2206–2218 CrossRef PubMed.
- A. M. Linsenmeier, C. M. Williams and S. Bräse, J. Org. Chem., 2011, 76, 9127–9132 CrossRef CAS PubMed.
- A. M. Linsenmeier, C. M. Williams and S. Bräse, Eur. J. Org. Chem., 2013, 3847–3856 CrossRef CAS PubMed.
- T. Caronna, S. Gabbiadini, A. Mele and F. Recupero, Helv. Chim. Acta, 2002, 85, 1–8 CrossRef CAS.
- S. V. Kessar, Y. P. Gupta, P. Singh, V. Jain and P. S. Pahwa, J. Chem. Soc. Pak., 1979, 1, 129–130 CAS.
- C. Bazzini, S. Brovelli, T. Caronna, C. Gambarotti, M. Giannone, P. Macchi, F. Meinardi, A. Mele, W. Panzeri, F. Recupero, A. Sironi and R. Tubino, Eur. J. Org. Chem., 2005, 1247–1257 CrossRef CAS PubMed.
- L. H. Klemm and A. Weisert, J. Heterocycl. Chem., 1965, 2, 140–143 CrossRef CAS PubMed.
- S. Djurdjevic, D. A. Leigh, H. McNab, S. Parsons, G. Teobaldi and F. Zerbetto, J. Am. Chem. Soc., 2007, 129, 476–477 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- L. E. Peisino and A. B. Pierini, J. Org. Chem., 2013, 78, 4719–4729 CrossRef CAS PubMed.
- A. B. Pierini and D. M. A. Vera, J. Org. Chem., 2003, 68, 9191–9199 CrossRef CAS PubMed.
- S. Miertus, E. Scrocco and J. Tomasi, J. Chem. Phys., 1981, 55, 117 CAS.
- S. Miertus and J. Tomasi, J. Chem. Phys., 1982, 65, 239–245 CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- D. B. G. Williams and M. Lawton, J. Org. Chem., 2010, 75, 8351–8354 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra04563k |
| ‡ In DMSO and DMF several concentrations of diamine 8 and base were explored, but the yield of cyclic products does not improve. |
| § For full optimization reaction see ESI.† |
| ¶ In the distonic specie, the negative charge is in π system meanwhile the radical is in σ system. |
| || The rotation barrier is ca. 17 kcal mol−1, from AM1 calculations. |
| ** The rotation barrier is ca. 23 kcal mol−1, from AM1 calculations. |
| †† http://www.verachem.com/products/vconf/. |
| This journal is © The Royal Society of Chemistry 2015 |