
Hydrothermal syntheses, structural characterizations, and magnetic properties of five MOFs assembled from C2-symmetric ligand of 1,3-di(2′,4′-dicarboxylphenyl)benzene with various coordination modes†
 *ab
*ab
Abstract
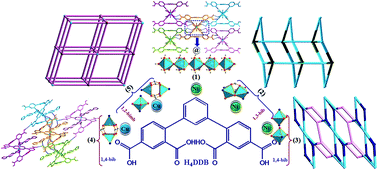
Five new complexes, namely, [Ni(H2DDB)(H2O)2(μ2-H2O)]n (1), [Ni1.5(DDB)(1,4-bib)1.5(H2O)]n (2), {[Ni2(DDB)(1,3-bib)2(μ2-H2O)]·2H2O}n (3), [Cu2(H2DDB)2(1,4-bib)2]·H2O (4), and {[Cu1.5(HDDB)(1,2-bimb)]·H2O}n (5), have been synthesized using the solvothermal reaction of 1,3-di(2′,4′-dicarboxylphenyl)benzene (H4DDB) with nickel(II) or copper(II) salts in the presence of ancillary ligands of bis(imidazole) linkers (1,4-bib = 1,4-bis(1H-imidazol-4-yl)benzene, 1,3-bib = 1,3-bis(1H-imidazol-4-yl)benzene, and 1,2-bimb = 1,2-bis(imidazol-1-ylmethyl)benzene). Their structures have been determined by single-crystal X-ray diffraction analyses and further characterized by elemental analyses, IR spectroscopy, powder X-ray diffraction (PXRD), and thermogravimetric (TG) analyses. In complex 1, NiII ions are bridged by associated water molecules to form an interesting [Ni(H2DDB)(H2O)2(μ2-H2O)]n chain structure, which is further extended to a 3D supramolecular structure via the hydrogen bonds. In complex 2, a novel 3D (3,3,6)-connected (63)4(65·88·102) net is generated from the synergistic effect of 1D [Ni(1,4-bib)]n zigzag chains and 2D [Ni3(DDB)2]n bilayers. For complex 3, with the employment of 1,3-bib bis(imidazole) linker instead of 1,4-bib, an unprecedented binuclear {Ni2(COO)(μ2-H2O)} SBUs based 3D (3,6)-connected (3·6·7)(32·43·54·63·7·82) net was obtained. Furthermore, when NiII ions were replaced by CuII ions, a paddle wheel {Cu2(COO)4} SBUs based [Cu2(H2DDB)(1,4-bib)2] (4) complex was formed. For complex 5, 2D [Cu3(HDDB)2]n sheets are further linked by 1,2-bimb pillars to expand a paddle wheel {Cu2(COO)4} SBUs based 3D architecture with 4-connected (65 8)-cds topology. Moreover, the magnetic properties of 1, 3, and 5 have been investigated.
 Please wait while we load your content...
Please wait while we load your content...

